Completion status #
Completed preps #
11/30/21 #
T7 initiation series transformations #
Using electrocompetent cells I prepared on 11/17 with Fred Tadas transformed most of the verified T7 init series mini preps, however a large number of the samples arced. To test if this was due to the cells or the preps themselves I zapped a sample with only the water used to dilute cells, and then with 1 ul of mini prep (same volume used for the transformations). While water alone did not arc the water + prep did indicating their is residual salt in some of the mini preps.
12/1/21 #
T7 initiation series transformations continued #
Picked up plates Tadas and I made yesterday this morning and most plates had > 25 colonies with some having hundreds.
Prepared 2L LB + 100 ug/ml Amp + 20 ml 20% glucose solution and divided into 6 1 L flasks with 300 ml per flask. Inoculated with one colony and incubated at 37C 200 rpm shaking starting at 3:00 pm on the second floor shaker.
12/2/21 #
T7 init midi prep preparation: standard protocol #
Followed lab mini prep protocol including reagent volumes. Spun cells down in 1 L flasks in the microbio centrifuge room then transfered to oakridge tubes. Difficult to resuspend but with lots of vortexing was able to. After SDS lysis lots of lysate was present. So much so that was difficult to remove fully and so I had to transfer to another oakridge tube and spin again. In future increase (double at least) the spin time when seperating lysis. Lysis was clear. Stopped at LiCl freeze and left in freezer overnight to finish in the morning.
12/3/21 #
VR n midi preps #
12/5/21 #
VR 6, 8, 9, 10, 11, 12 midi preps #
Cultured individual colonies in 250 ml 1x LB 0.2 % glucose and 100 ug/ml amp.
12/6/21 #
Gels to visualize midi prep results. In both gels using pFC9 as a loading control. Both gels used 1x TAE in agarose with 1ul/100ml TAE EtBr in agarose and running buffer. All laders are 1kb GeneRuler.
Midi prep gels show plasmid recombination #
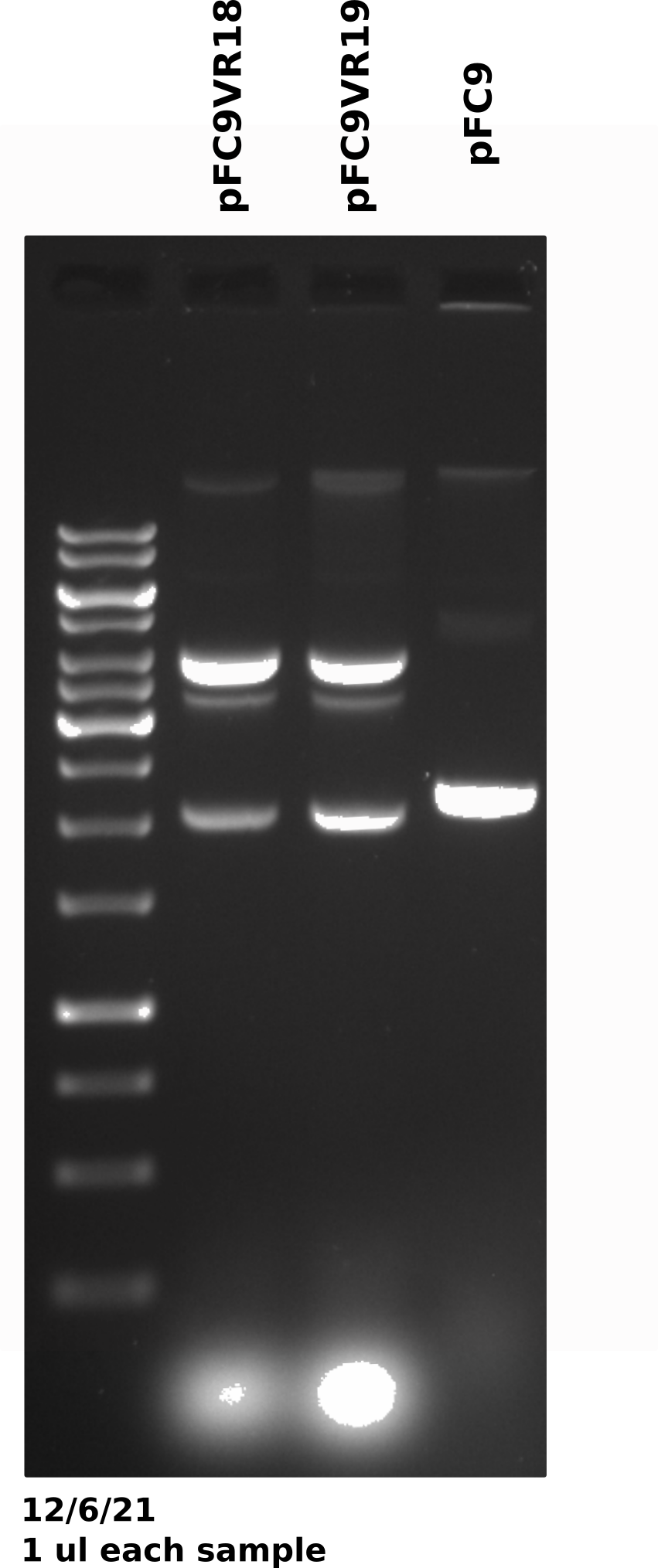
pFC9VR8-11 gel #
Gel was run for 60V for 20 mins then voltage was increased to 130 for another 20 mins (I was trying to get out of lab).
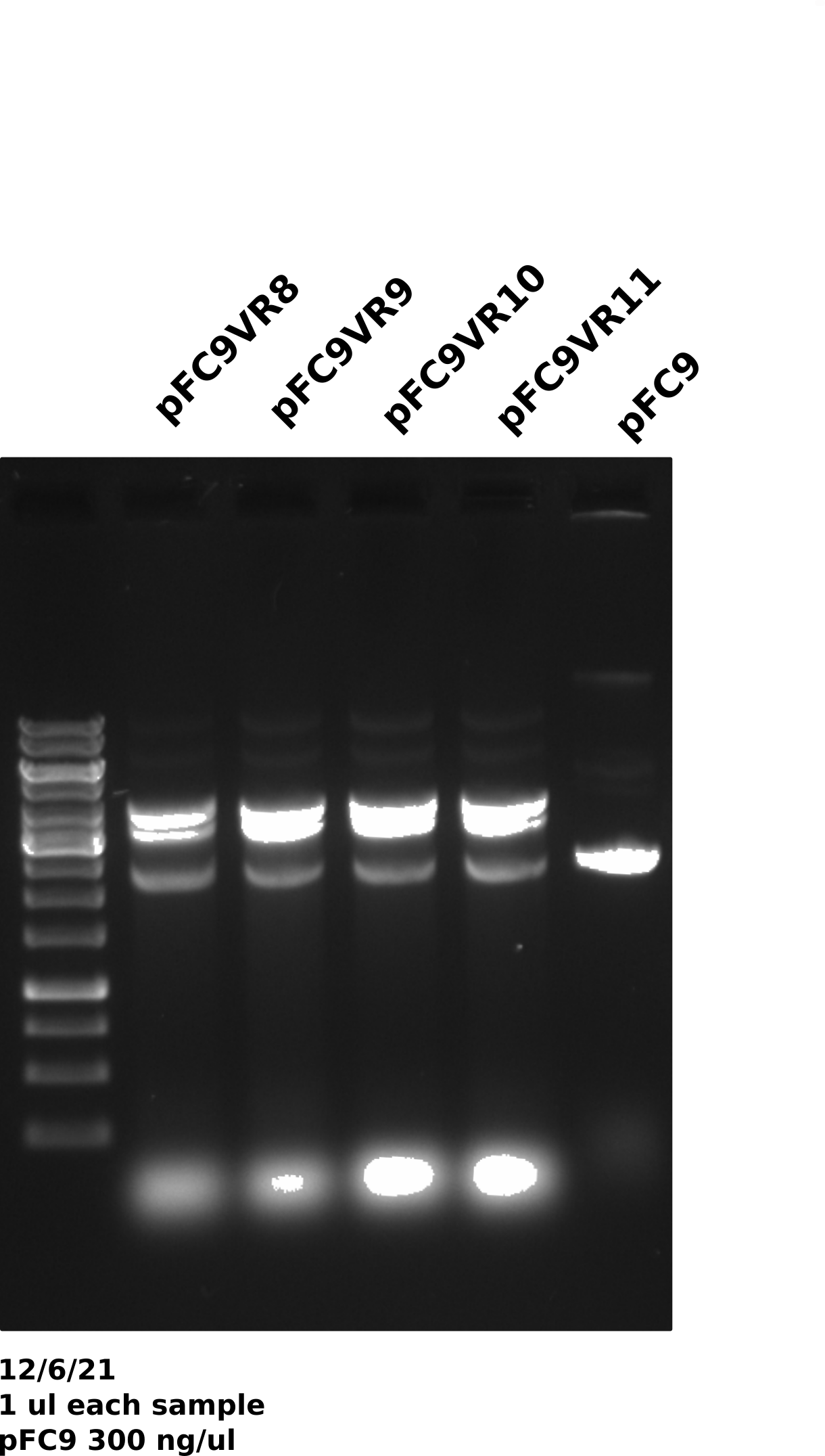
Duplex band is present across preps (done on different days) and across plasmids which may be indicating that growth conditions may be affecting plasmid recombination. The fact that this was not an issue in mini preps also points to this.
12/7/21 #
Plasmid recombination issues and possible solutions #
Talked with Fred about recombination issues with previous midi preps. Since mini preps do not show same issues, the next place to look would be growth conditions. Today will first digest previous preps to linearize and confirm recombination is occurring and then set up series of growth conditions with one plasmid and midi prep them tomorrow to see if can affect recombination by changing grow conditions.
T7 init midi prep EcoRI digest #
Digesting 8 of midi preps I have done so far with EcoRI to linearize to determine if duplex band is destroyed by digestion. For each sample added 2 ul plasmid, 2ul 2.2 10x buffer and 14 ul water. Then split into 2 10 ul aliquotes and added 0.5 ul EcoRI to the digested sample. All samples incubated for 1 hour at 37C in the thermocycler. Samples and gel layout are listed in the tables below.
Digested samples #
| Insert | Date |
|---|---|
| 8 | 12/06/21 |
| 9 | 12/06/21 |
| 10 | 12/06/21 |
| 11 | 12/06/21 |
| 13 | 12/05/21 |
| 17 | 12/05/21 |
| 18 | 12/05/21 |
| 19 | 12/05/21 |
Gel layout #
| Lane | Sample | Condition |
|---|---|---|
| 1 | 1kb ladder | NA |
| 2 | VR8 | Control |
| 3 | VR8 | EcoRI |
| 4 | VR9 | Control |
| 5 | VR9 | EcoRI |
| 6 | VR10 | Control |
| 7 | VR10 | EcoRI |
| 8 | VR11 | Control |
| 9 | VR11 | EcoRI |
| 10 | VR13 | Control |
| 11 | VR13 | EcoRI |
| 12 | VR17 | Control |
| 13 | VR17 | EcoRI |
| 14 | VR18 | Control |
| 15 | VR18 | EcoRI |
| 16 | VR19 | Control |
| 17 | VR19 | EcoRI |
| 18 | 1kb ladder | NA |
| 19 | 1kb ladder | NA |
Ran for 2 hours at 60V. 0.8% 1x TAE agarose and buffer 1 ul/ml EtBr.
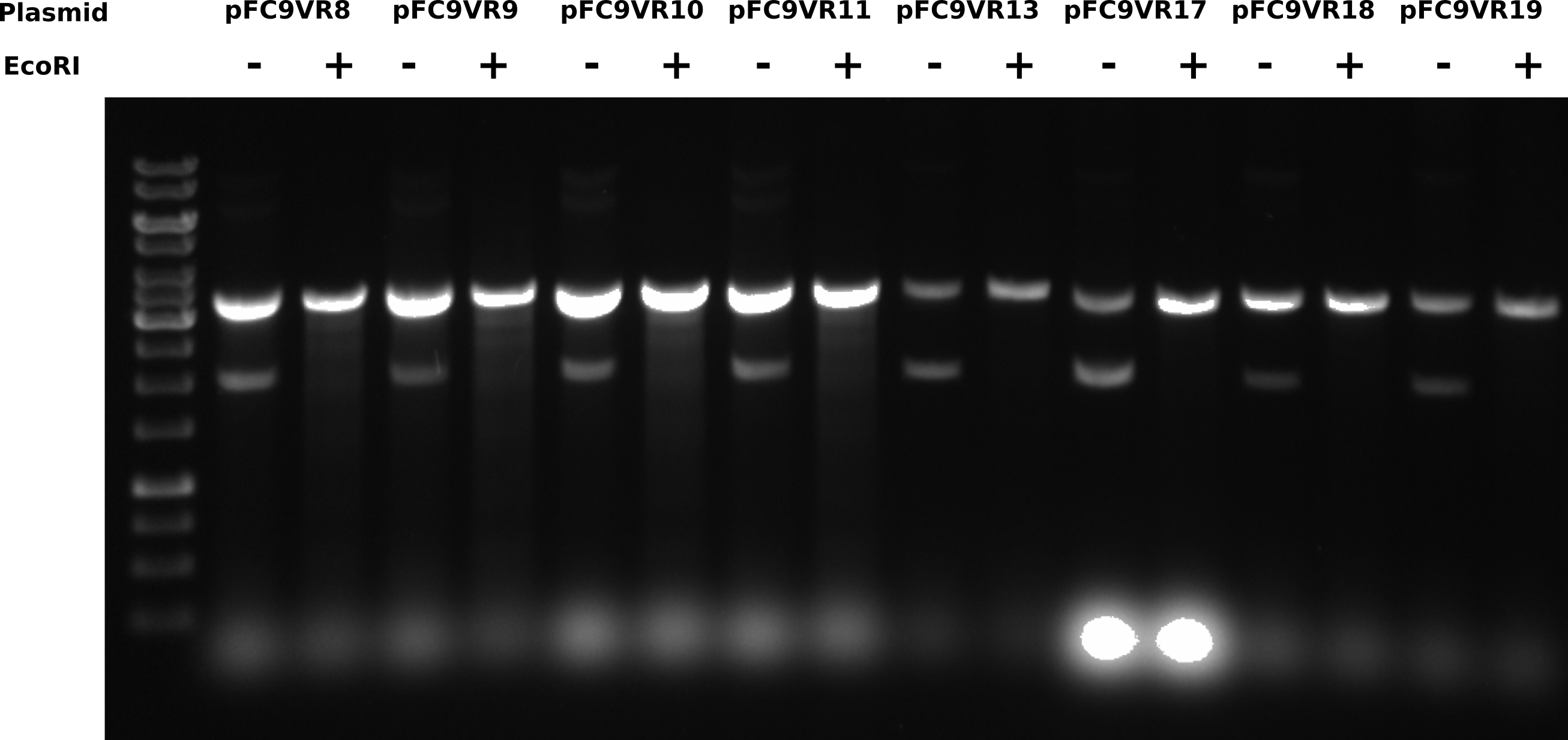
Across all samples, digesting with EcoRI shifts bottom band to same height as the top band. Where is the nicked band that is seen in previous gels from the 6th?
A note on the effect of salts on DNA migration
After talking to Fred about this gel he mentioned that salt can have strong effects on DNA migration patterns. All samples in this gel would have had additional salt from the restriction buffer. This may explain why the nicked band is not visible in the control samples as it could have been effected by salt placing it at the same height of duplex plasmids. Either way the supercoiled DNA population is the minority in all of these samples.
Effect of growth conditions on plasmid recombination: Experimental setup #
Table of tested conditions #
Testing 13 conditions as that is close to max number of midi preps I can do in a day given the number of centrifuge bottles I have available.
| Sample number | Flask volume (L) | % glucose | culture volume | temperature (C) |
|---|---|---|---|---|
| 1 | 1 | 0 | 250 | 30 |
| 2 | 1 | 0.2 | 250 | 30 |
| 3 | 1 | 0 | 500 | 30 |
| 4 | 1 | 0.2 | 500 | 30 |
| 5 | 1 | 0 | 150 | 30 |
| 6 | 1 | 0.2 | 150 | 30 |
| 7 | 1 | 0 | 150 | 37 |
| 8 | 1 | 0 | 500 | 37 |
| 9 | 1 | 0.2 | 150 | 37 |
| 10 | 0.5 | 0 | 250 | 37 |
| 11 | 0.5 | 0.2 | 250 | 37 |
| 12 | 4 | 0.2 | 350 | 37 |
| 13 | 4 | 0 | 350 | 37 |
Started samples at their respective temperatures in the second floor shaker room at 4 PM. All samples shaking at 200 rpm. All samples inoculated with 1 colony from pFC9VR18 midi prep plate.
12/8/21 #
Effect of growth conditions on plasmid recombination: Experiment execution #
Picked up samples from shakers around 9:00 AM; negative control was negative indicating contaimination was not a problem. I began midi prepping all samples at 9:20. Stopped after the isopropanol precipitation due to time limitations.
These samples are not destined for anything other than a gel so if they are dirty street plasmids so be it. I was spinning down lysis right before NSF R-loop group meeting and so I did not have time to spin samples for the full 30 mins that I recommended previously and so some samples still retained a fair amount of lysis precipitate that was evident when re-suspending in 10 mM Tris Hcl after 2 EtOH washes. After re-suspending (0.5 ml) I spun all samples for 13 min at 15k rpm and then transferred supernantent to a new tube in order to remove any lysis crap that remained in the samples.
Agarose gel #
Ran all samples on gel 0.8% agarose 1x TAE 1ul/ml EtBr in gel and buffer ran for 60V for 2 hours.

12/9/21 #
Effect of growth conditions on plasmid recombination: Experiment analysis #
First I quantified the gel I ran yesterday using the ImageLab software on the lab computer. I exported the quantification’s and did all analysis in this Jupyter notebook.

I calculated the ratio of percent lane signal in the supercoiled band over the ratio percent lane signal in the nicked / duplex band for each condition. This is quantified in the plot below and shown with sample conditions in the table.
Supercoiling ratio all samples #
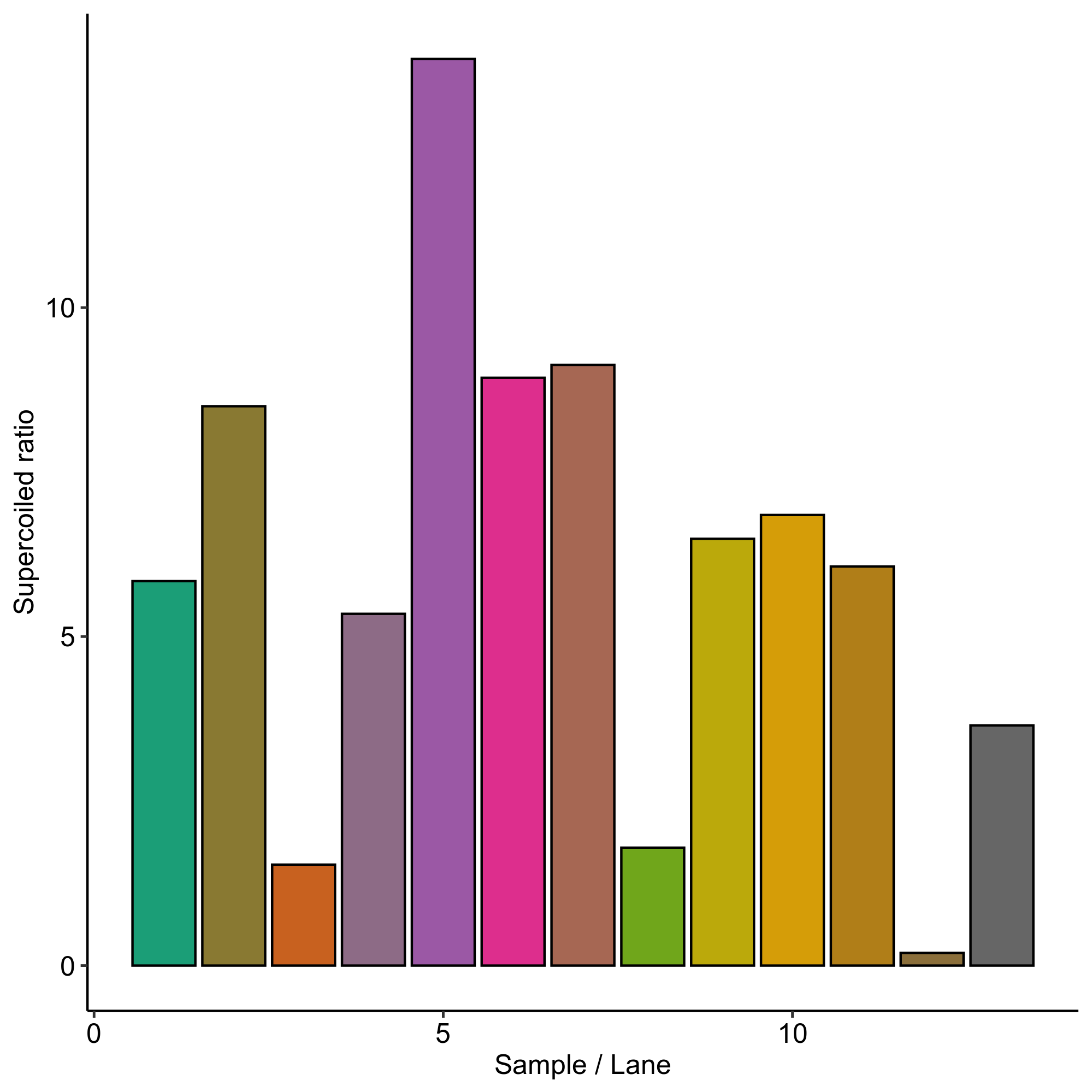
Sample table with supercoiling ratios #
| Lane | Flask volume (L) | % glucose | culture volume | temperature (C) | Supercoiling ratio |
|---|---|---|---|---|---|
| 1 | 1 | 0 | 250 | 30 | 5.84288 |
| 2 | 1 | 0.2 | 250 | 30 | 8.50091 |
| 3 | 1 | 0 | 500 | 30 | 1.53411 |
| 4 | 1 | 0.2 | 500 | 30 | 5.3459 |
| 5 | 1 | 0 | 150 | 30 | 13.7793 |
| 6 | 1 | 0.2 | 150 | 30 | 8.93258 |
| 7 | 1 | 0 | 150 | 37 | 9.12953 |
| 8 | 1 | 0 | 500 | 37 | 1.79163 |
| 9 | 1 | 0.2 | 150 | 37 | 6.48647 |
| 10 | 0.5 | 0 | 250 | 37 | 6.84805 |
| 11 | 0.5 | 0.2 | 250 | 37 | 6.06537 |
| 12 | 4 | 0.2 | 350 | 37 | 0.193021 |
| 13 | 4 | 0 | 350 | 37 | 3.6497 |
Sample comparisons across conditions #
150 ml volume #
When grown at 30C without glucose saw best supercoiled ratio. But unlike other superoiled samples glucose decreased supercoiled ratio. Glucose also decreased supercoiling ratio when grown at 37C.
500 ml volume #
Glucose #
When grown at 30C glucose also increased relative supercoiled percentage. Similar increase was also seen when glucose added in the same volume of culture but grown at 37C.
Temperature #
At 500ml no glucose increasing temperature to 37 slightly increased ratio but basically no effect.
Conclusions #
Overall I would say I trust this data maybe 30%. It is overall very noisy but seems to suggest that the best growth conditions would be with minimal O2. Another theory is that all of these conditions are noisy and the main factor is actually the individual colony picked. However this would mean I happened to pick almost all decent colonies on this go around but almost all bad colonies of the first midis.
After talking with Fred I decided the best way forward would be to test the best two conditions with a few replicates each; all from the same plasmid but different colonies and midi these samples. This tests
- Sweet spot hypothesis: There are some growth conditions that are just better at getting supercoiled DNA than others.
- Bad colony hypothesis: Growth condition factors are outweighed by the bacterial starting material.
Effect of growth conditions on plasmid recombination: Best condition reproducibility #
Testing best conditions from yesterday with multiple samples from the same plate. This will confirm if these conditions can be used for further preps and if data is reproducible between replicates and experiments.
Sample table #
| Sample number | Flask volume (L) | % glucose | culture volume | temperature (C) | Plasmid |
|---|---|---|---|---|---|
| 1 | 1 | 0 | 150 | 30 | VR18 |
| 2 | 1 | 0 | 150 | 30 | VR18 |
| 3 | 1 | 0 | 150 | 30 | VR18 |
| 4 | 1 | 0 | 150 | 37 | VR18 |
| 5 | 1 | 0 | 150 | 37 | VR18 |
| 6 | 1 | 0 | 150 | 37 | VR18 |
| 7 | 1 | 0 | 500 | 37 | VR18 |
| 8 | 1 | 0 | 500 | 37 | VR18 |
Cultures #
Started cultures at respective temperature at 4:30 PM second floor shakers at 200 rmp. All samples using LB I autoclaved earlier today, allowed to cool and then added 100 ug/ml Amp.
12/10/21 #
Effect of growth conditions on plasmid recombination: Best condition reproducibility execution #
Picked up cultures from shakers at 8:30 am and got to work on the midi preps. Transferred all cultures to 500 ml autoclaved centrifuge bottles. Did not use full volume of samples 7 and 8. Spun down in SLA3000 re-suspended, lysed and neutralized pellets using Tada’s solutions I, II, and III. After re-suspension transferred samples 7 and 8 into 250 ml centrifuge bottles so could spin in JLA 16-250 rotor and the rest of the samples in SLA3000. To separate lysis from supernatent spun all samples 10k rpm for 15 minutes. After spin removed supernanent and transferred all samples to Oakridge tubes. After transfer spun all samples in the JA-14-50 rotor for 12 mins (takes about 2 mins for rotor to get up to speed) at 14k rpm 4C.
Agarose gel #
Gel conditions #
Ran all samples on 0.8 % agarose 1x TAE (buffer and agarose) 1ul/100ml EtBr gel at 60V for 2 hours.
Lane layout #
| Lane | Sample | Date |
|---|---|---|
| 1 | 1 kb ladder | NA |
| 2 | 1 | 12/10/21 |
| 3 | 2 | 12/10/21 |
| 4 | 3 | 12/10/21 |
| 5 | 4 | 12/10/21 |
| 6 | 5 | 12/10/21 |
| 7 | 6 | 12/10/21 |
| 8 | 7 | 12/10/21 |
| 9 | 8 | 12/10/21 |
| 10 | 5 | 12/08/21 |
| 11 | 7 | 12/08/21 |
| 12 | 8 | 12/08/21 |
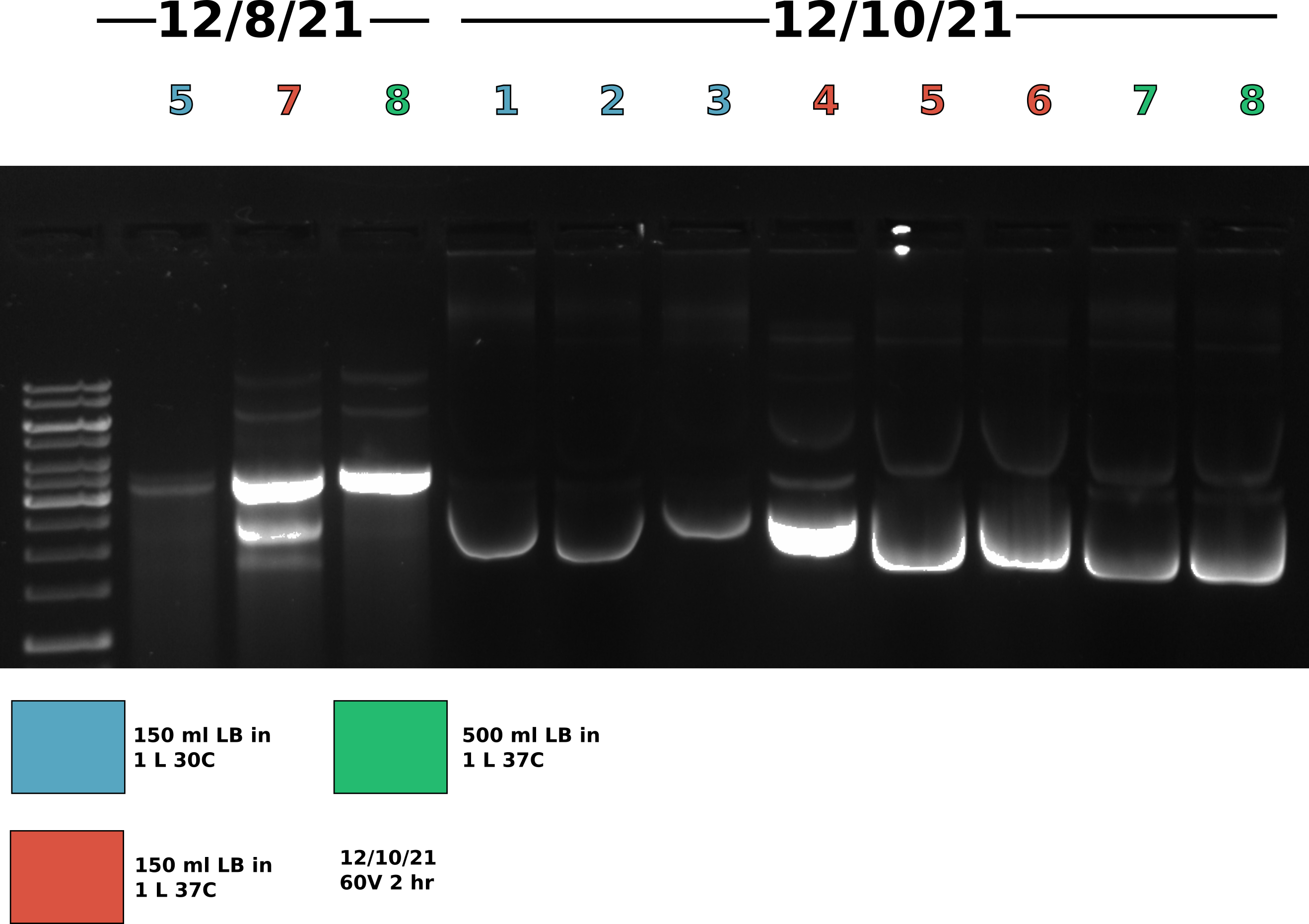
While band shape is very strange gel does seem to show consistency across conditions and colonies. Going to move forward with growing 150 ml of culture in 1 L flask at 37C as this gives good yields and minimal amount of dimer. THe control lanes (samples from 12/10/21) do not look like gel run on same day the preps were completed likely due to the fact these preps were very dirty and left at room temp overnight. Proteins still in solution (def Dnase in sample 1) were likely still active.
12/11/21 #
Set up for midi preps: 17, 19 20-23, 25-30 #
Prepared 3 L 1x LB and dispensed 150 ml into 12 different 1 autoclaved 1 L flasks. Inoculated each flask with 1 colony using pipette tip and places flasks on second floor shaker at 200 rpm 37C.
Prepping colonies from inserts 17, 19, 20, 21, 22, 23, 25, 26, 27, 28, 29 and 30.
12/12/21 #
Midi prep 17, 19 20-23, 25-30: execution #
Picked up cultures at 8:40 AM and brought to LSA third floor centrifuge room along with midi prep materials and started preps around 9 AM. All innoculated flasks grew and negative control showed no signs of growth.
Negative control

All cultures after removing from shaker

Completed LiCl clean up around 3 pm and re-suspended samples in 0.5 ml of 10 mM Tris Hcl. Placed all samples in freezer.
12/13/31 #
Midi prep 17, 19 20-23, 25-30: agarose gel #
Prepared agarose gel to visualize midi preps I completed with Tadas yesterday. Loaded 1 ul each sample with 9 ul npH20 (5ml Dnase Rnase free aliquots) with 1.8 ul loading dye. Loaded 10 ul each sample using multichannel pipette into wells.
Gel conditions #
Ran all samples on 0.8 % agarose 1x TAE (buffer and agarose) 1ul/100ml EtBr gel at 60V for 2 hours. TAE was made fresh this morning from 50x concentrate.
Sample layout #
| Lane | Sample |
|---|---|
| 1 | pFC9VR17 |
| 2 | pFC9VR19 |
| 3 | pFC9VR20 |
| 4 | pFC9VR21 |
| 5 | pFC9VR22 |
| 6 | pFC9VR23 |
| 7 | pFC9VR25 |
| 8 | pFC9VR26 |
| 9 | pFC9VR27 |
| 10 | pFC9VR28 |
| 11 | pFC9VR29 |
| 12 | pFC9VR30 |
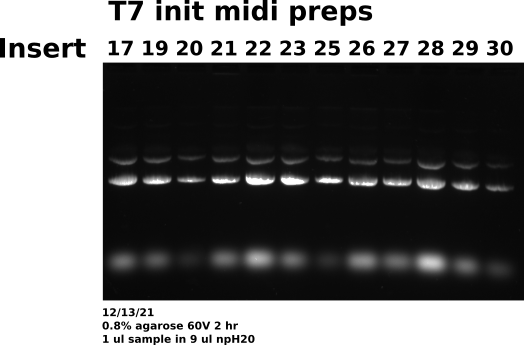
Midi prep samples VR 1-3, 4-13: Preparation #
Aliquoted LB with 100 ug / ml Amp into 1 L flasks; 150 ml LB per flask. Then inoculated with 1 colony of each verified plasmid prep transformation using pipette tip. Placed all samples on second floor shakers at 200 rpm at 37C to shake overnight.
12/14/21 #
Midi prep samples VR 1-3, 4-13: Execution #
Picked up samples at 6:45 AM this morning. Negative control was negative but sample pFC9VR7 did not grow well. Cell density was clearly significantly lower than all other samples. Continued with it in the preps mainly to maintain an even number of samples for centrifuging. Carried out midi prep and LiCl prepcipitation as normal. Re-suspedended all samples in 0.5 ml 10mM Tris HCl.
Agarose gel #
Gel conditions #
0.8 % agarose gel made with 1x TAE in agarose and running buffer. 1 ul / 100 ml EtBr in agarose and running buffer. Ran gel at 60V for 2 hours.
Well layout #
| Lane | Sample |
|---|---|
| 1 | 1kb ladder |
| 2 | pFC9VR-1 |
| 3 | pFC9VR-2 |
| 4 | pFC9VR-3 |
| 5 | pFC9VR-5 |
| 6 | pFC9VR-6 |
| 7 | pFC9VR-7 |
| 8 | 1kb ladder |
| 9 | pFC9VR-8 |
| 10 | pFC9VR-9 |
| 11 | pFC9VR-10 |
| 12 | pFC9VR-11 |
| 13 | pFC9VR-12 |
| 14 | pFC9VR-13 |
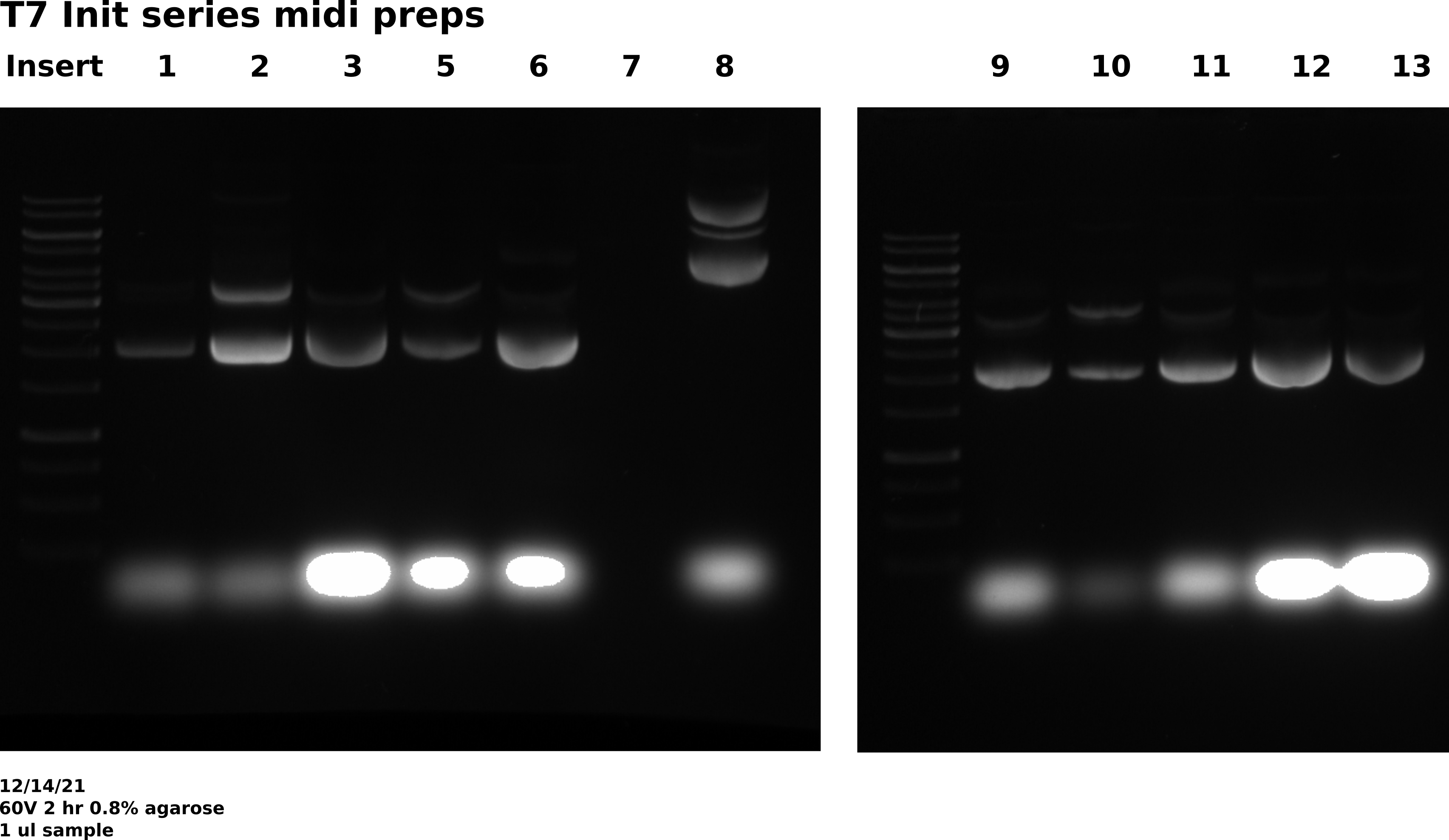
As expected there is basically no DNA in pFC9VR7 sample but unexpectedly it looks like whatever was in pFC9VR8 was not my plasmid. All other samples look as expected. Will redo 7 and 8 on Thursday preps.
pFC9VR4 + Talysa plasmid transformations #
There was visible contamination on the pFC9VR4 plate so in preparation for Thursday midi preps I retransformed using 12/1/21 homemade chemically competent cells using 1 ul of plasmid and following standard NEB transformation protocol. I also transformed 2 plasmids for Talysa in the same fashion; RnaseH wildtype and mutant vectors. Plated 1/4th total volume of each transformation (250 ml) into LB Amp plates and placed into 37C room to grow overnight.
12/15/21 #
pFC9VR4 + Talysa plasmid transformations: results #
Picked up plates from 37C room around 9:10 AM. Both negative controls were negative.
![]()
Midi prep sequencing round 1 #
Sending in current midi preps which have promising gel results for sanger sequencing to verify their identify and make sure their traces look contamination free.
Sample prep #
- 5 ul each plamid prep
- 2.5 ul pFC9_T7_primer_1
- 7.5 ul npH20
Sample labeling #
| Sample number | Plasmid | Midi date |
|---|---|---|
| 1 | pFC9VR1 | 12-14-21 |
| 2 | pFC9VR2 | 12-14-21 |
| 3 | pFC9VR3 | 12-14-21 |
| 4 | pFC9VR5 | 12-14-21 |
| 5 | pFC9VR6 | 12-14-21 |
| 6 | pFC9VR9 | 12-14-21 |
| 7 | pFC9VR10 | 12-14-21 |
| 8 | pFC9VR11 | 12-14-21 |
| 9 | pFC9VR12 | 12-14-21 |
| 10 | pFC9VR13 | 12-14-21 |
| 11 | pFC9VR17 | 12-12-21 |
| 12 | pFC9VR19 | 12-12-21 |
| 13 | pFC9VR20 | 12-12-21 |
| 14 | pFC9VR21 | 12-12-21 |
| 15 | pFC9VR22 | 12-12-21 |
| 16 | pFC9VR23 | 12-12-21 |
| 17 | pFC9VR25 | 12-12-21 |
| 18 | pFC9VR26 | 12-12-21 |
| 19 | pFC9VR27 | 12-12-21 |
| 20 | pFC9VR28 | 12-12-21 |
| 21 | pFC9VR29 | 12-12-21 |
| 22 | pFC9VR30 | 12-12-21 |
T7 init 4, 7, 8, 14, 15, 16, 18, 24 midi preps: Preparation #
Prepared 150 ml LB + amp per insert and inoculated with 1 colony from each plate. All samples grown in 1 L flask. Placed flasks in second floor shakers 200 rpm 37C at 4:30 pm.
12/16/21 #
T7 init 4, 7, 8, 14, 15, 16, 18, 24 midi preps: Execution #
Picked up cultures at 8:30 and started midi prep protocol. Completed LiCl
purification around 3:00 PM. Re-suspended all samples in 0.5 ml 10mM Tris-HCl.
Talysa needed two plasmids prepped so she helped out and included two of
her own samples RnaseH1 WT and RnaseH1 Mut plasmids.
Midi prep agarose gel #
Gel conditions #
0.8% 1x TAE agarose gel 1ul/100 ml EtBr in gel and running buffer. Ran gel at 120V for 45 minutes.
Gel layout #
| Lane | Sample |
|---|---|
| 1 | 1kb ladder |
| 2 | pFC9VR4 |
| 3 | pFC9VR7 |
| 4 | pFC9VR8 |
| 5 | pFC9VR14 |
| 6 | pFC9VR15 |
| 7 | pFC9VR16 |
| 8 | pFC9VR18 |
| 9 | pFC9VR24 |
| 10 | 1kb ladder |
| 11 | RnaseH1 WT |
| 12 | RnaseH1 Mut |
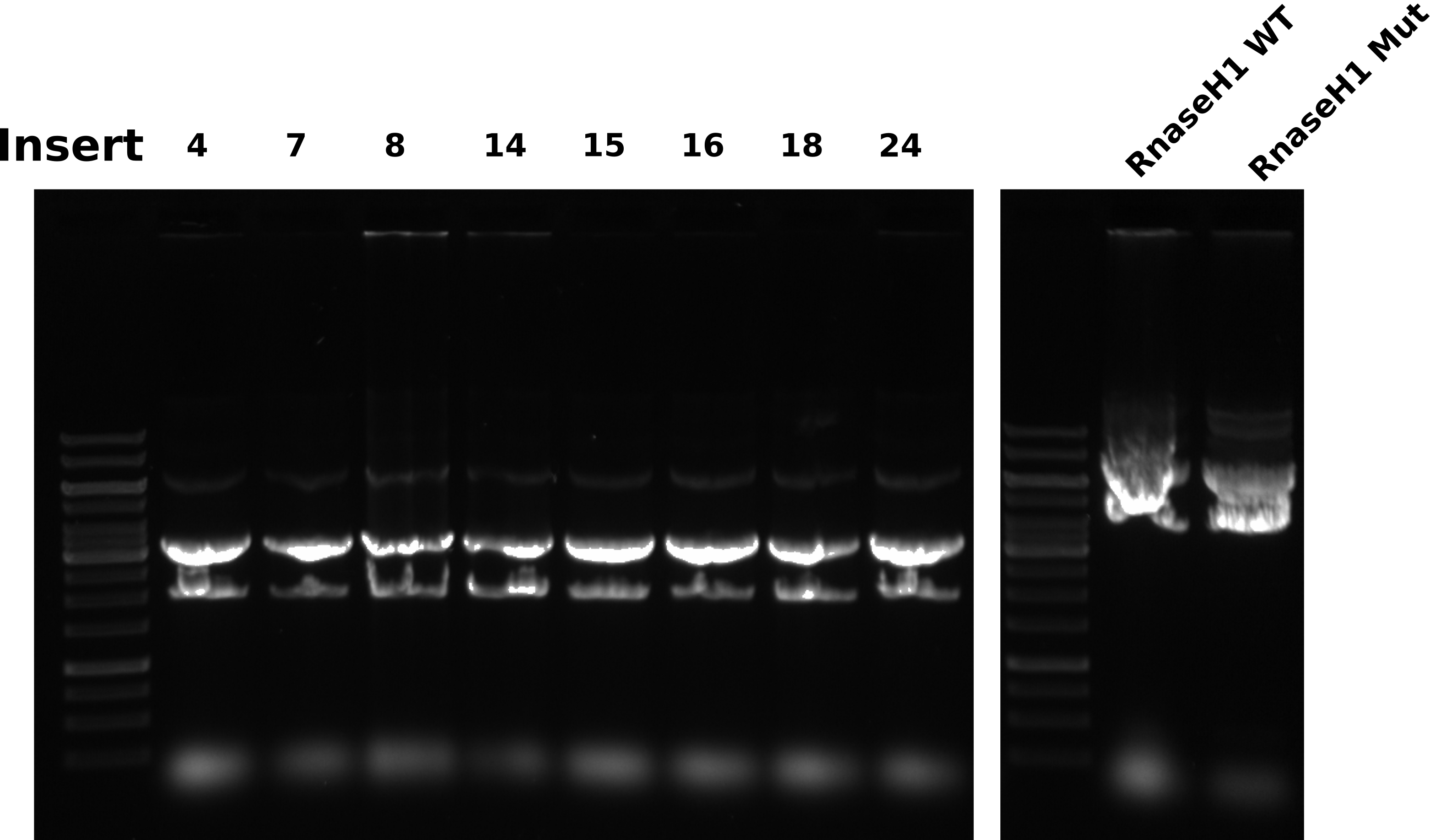
Bands are all really wacky shapes while the ladder ran fine. I have been seeing this across midi prep samples so it seems likely that there is something being left behind in the samples that is causing bands to run strangely. Maybe residual EtOH?. These samples were vacuumed multiple times and allowed to air dry for around an hour. It could be possible that an overnight dry is needed. Either way, it looks like preps will have to be re-done for these samples. All growth conditions were the same as previous preps, but sure what is changing that is effecting all preps in the same way.
Amp induced Dimer catasphroe hypothesis #
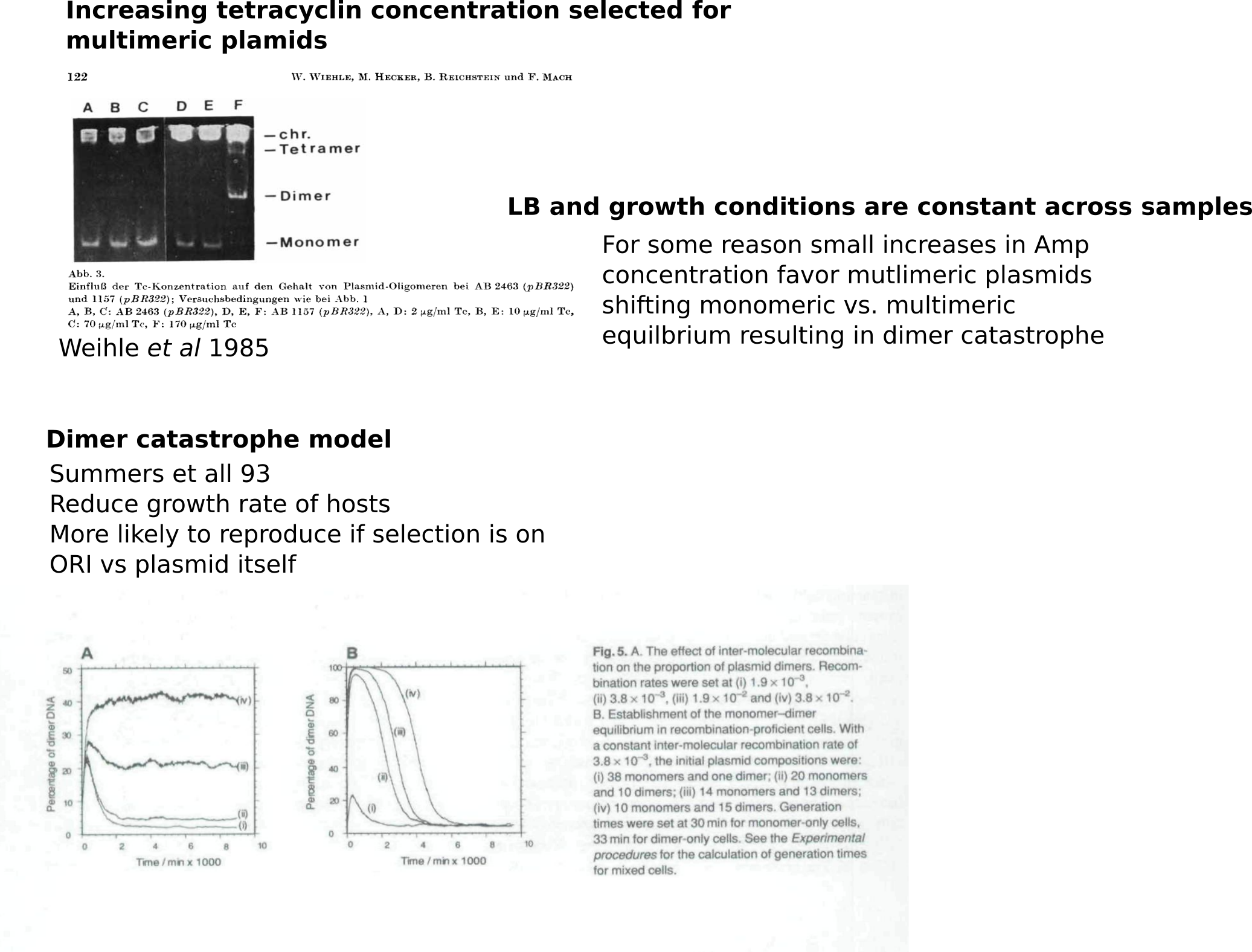
Midi prep sequencing round 1: results #
Got Sanger results returned from Quintara. Results can be downloaded from this link. Plot taken from report generated using the VR-verify workflow.
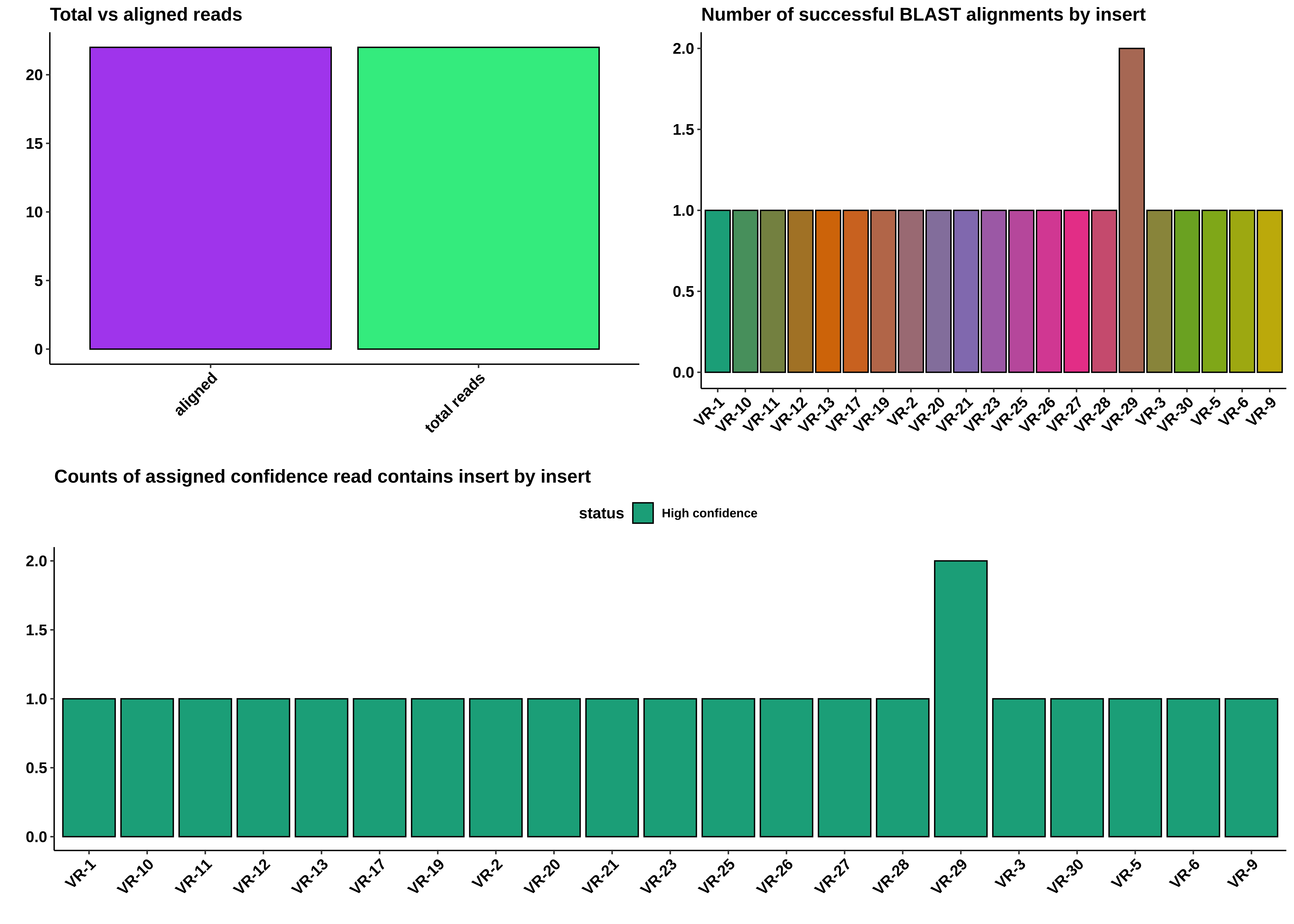
Overall results look good. Main issue is that there are two alignments to VR-29 so will have to redo the VR22 sample. All other samples aligned to expected insert with good quality scores.
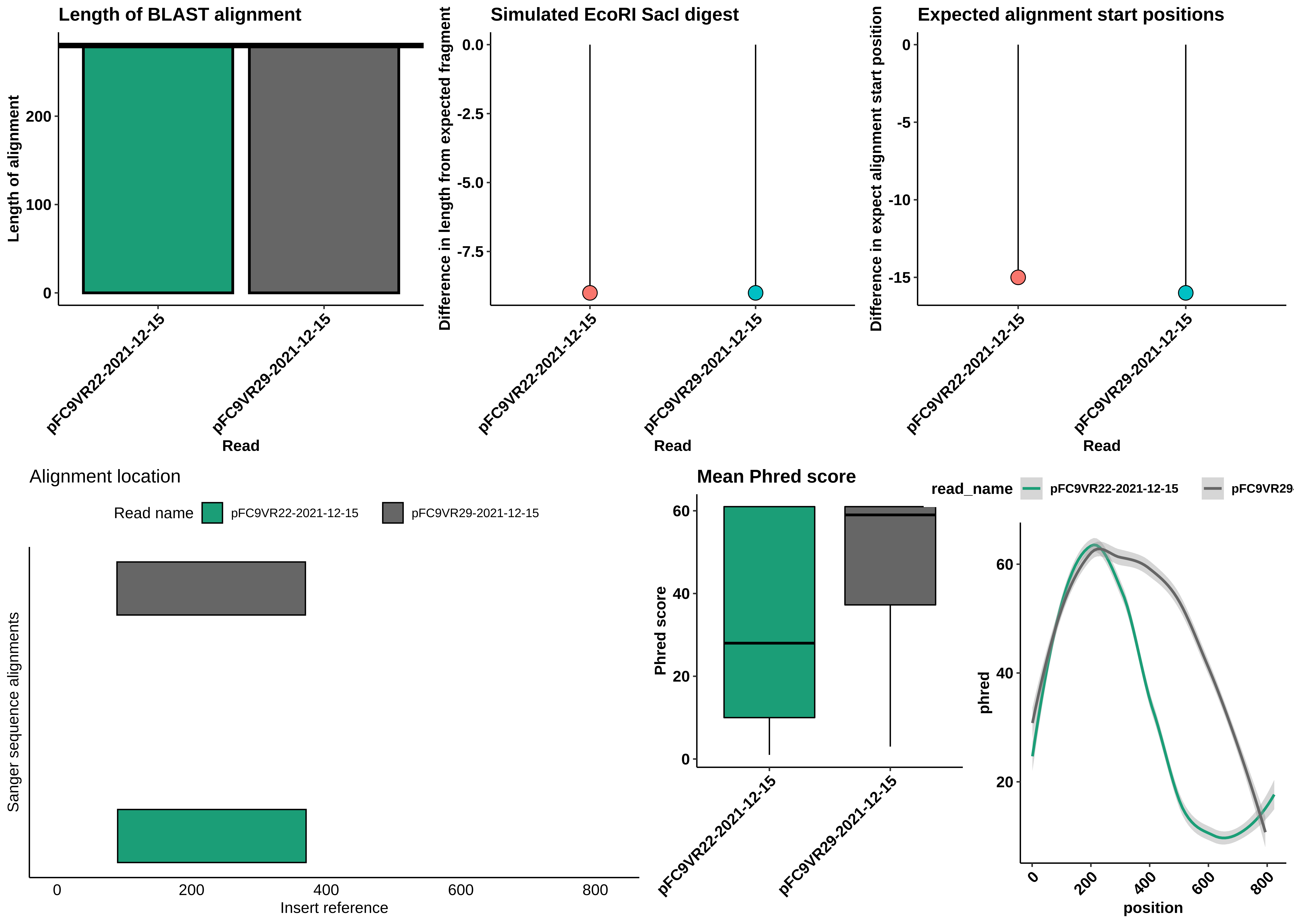
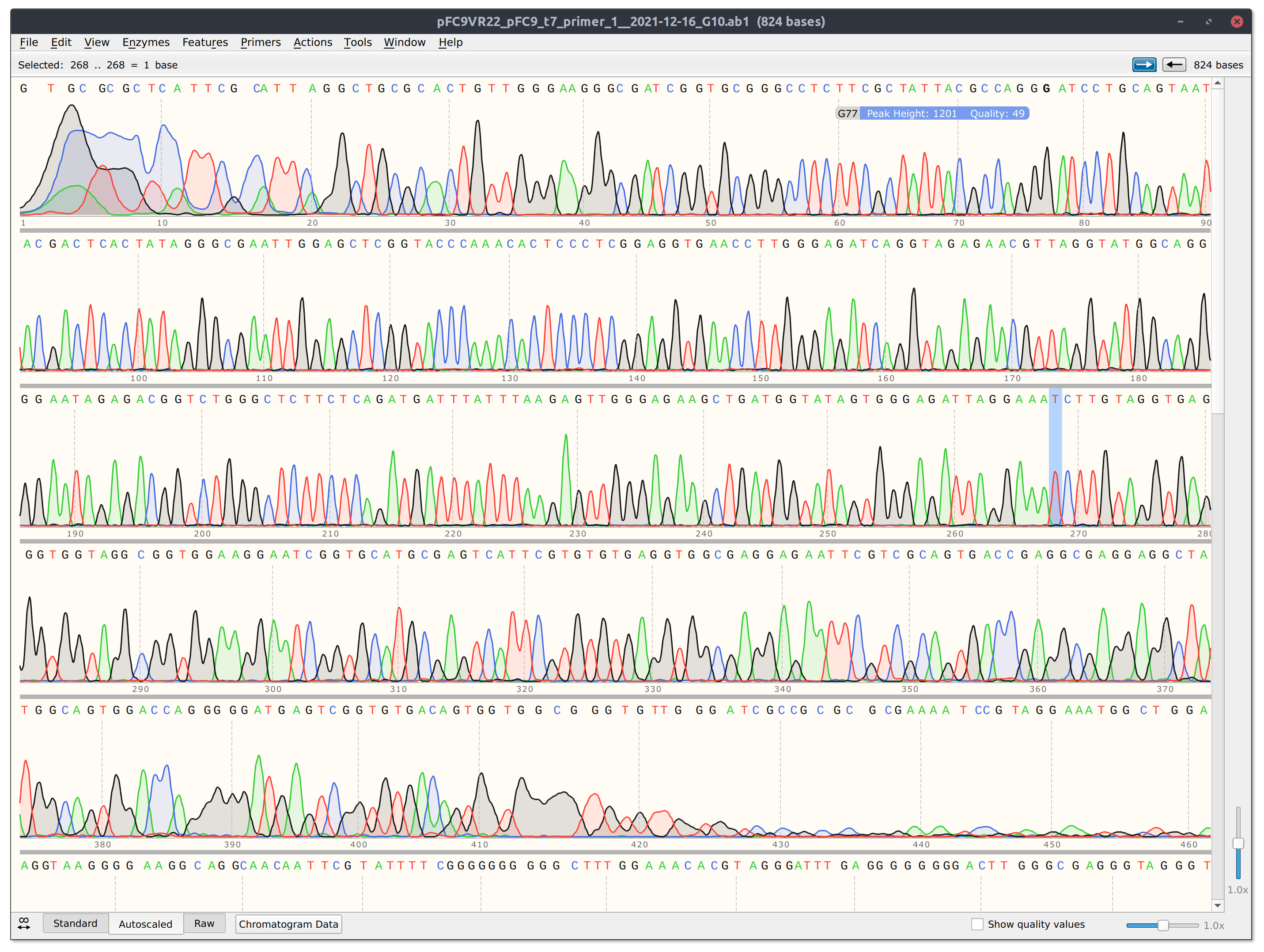
However looking at the trace for VR22 (shown in screenshot below) the read is pretty short and dies at a GC rich stretch so it may be worth sequencing from the other direction.
12/17/21 #
Re-run T7 init 4, 7, 8, 14, 15, 16, 18, 24 midi preps gel #
Since bands looked so wacky in previous gel I re-ran but at a much lower voltage; 45V for 3.5 hours. Gel layout and conditions (besides voltage) are the same as the 12/17 gel. 1 ul of each sample was used (also same as 12/17 gel).
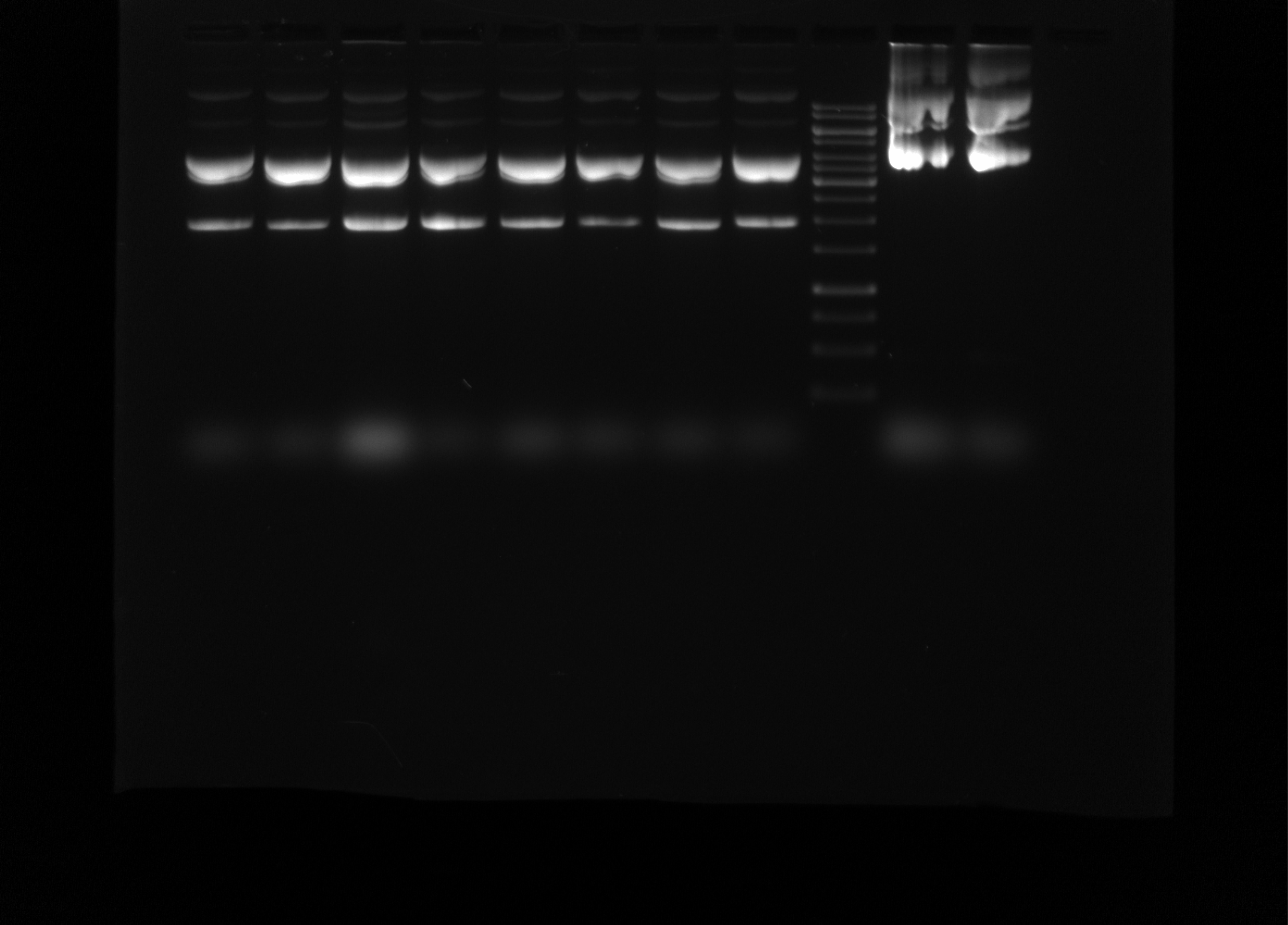
Overall the bands look much cleaner, indicating that the problem was likely just the preps have very high concentrations of DNA that was being run too fast.
12/19/21 #
Midi prep T7 init 4, 7, 8, 14, 15, 16, 18, 22, 24, 31 round II: Preparation #
Preparing for midi prep tomorrow. Repeating 12/17/21 since all samples from that prep were mostly dimers. Adding VR31 and 22.
LB preparation #
Prepared 2L 1x LB in 2L graduated cylinder. Then aliquoted to 2 1 L flasks and autoclaved with 6L water in tray in autoclave 2 for 35 minutes with no exhust time. After autoclaving LB was allowed to cool to room temperature over several hours before adding 0.1g of AMP to each 1L flask (100 ug / ml final concentration).
12/20/21 #
Midi prep T7 init 4, 7, 8, 14, 15, 16, 18, 22, 24, 31 round II: Execution #
Picked up cultures at 8:30 AM this morning. Negative control flask showed no growth while all other cultures grew. Started midi protocol around 9:00 AM and completed Isopropanol precipitation around 11:00. Tried pellets for 1 hour and re-suspended in 0.5 ml 10 mM Tris HCl.
Agarose gel #
0.8% 1x TAE gel, ran 60V for 2 hours. Loaded 0.5 ul each sample and 2.5 ul ladder.
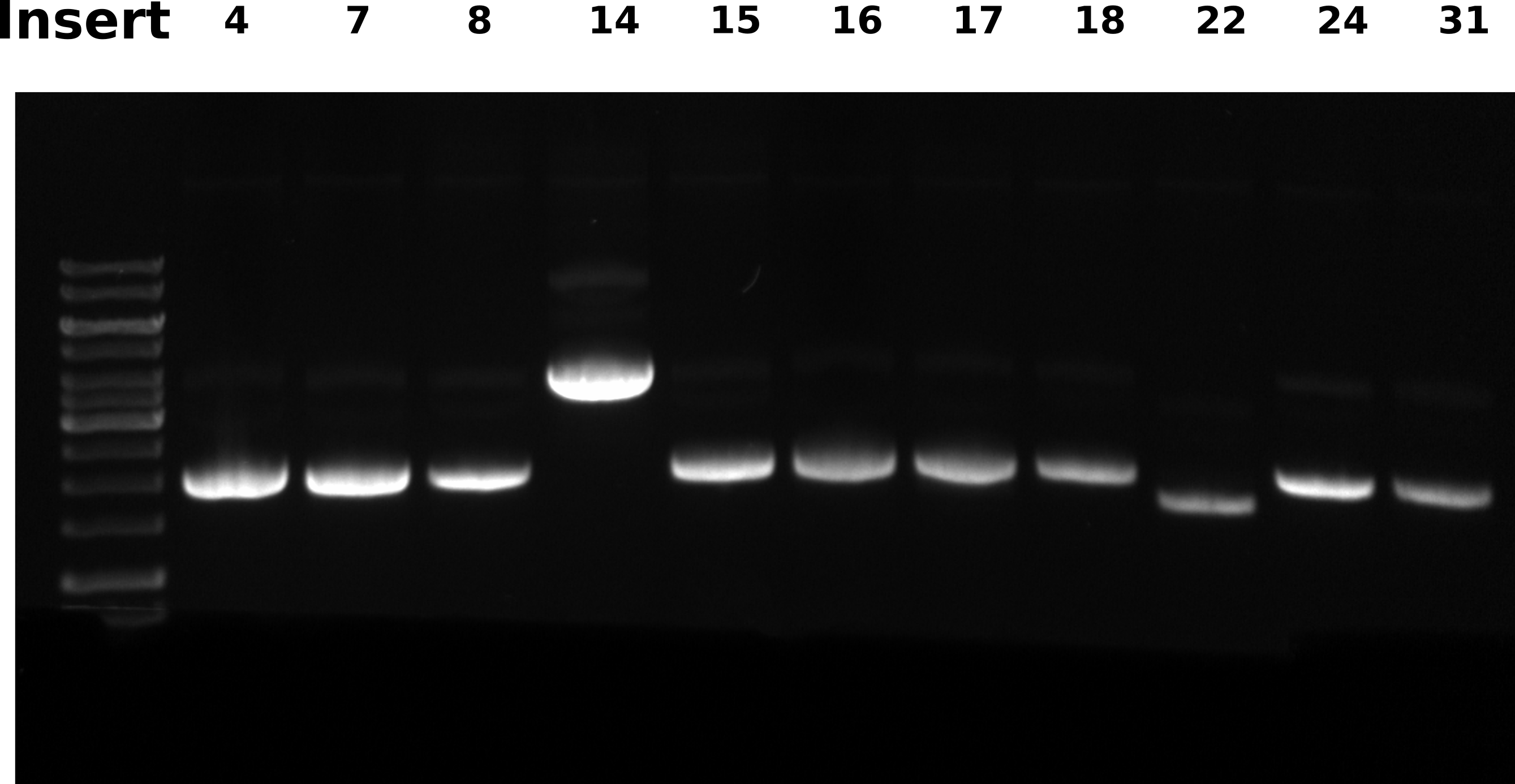
All samples have RNA contamination, I sliced the gel to remove the RNA “band” to make DNA bands more visible in this image.
12/21/21 #
Midi prep T7 init 4, 7, 8, 14, 15, 16, 18, 22, 24, 31 round II: Finishing up #
Completed LiCl clean up for with all samples. Re-suspended in 500 ul 10 mM Tris HCl and stored in freezer.
Transformations #
Based on gel from yesterday it looked like VR14 and VR22 need to be re-done. The VR7 plate as no more colonies and the identity of VR22 is in question. Did standard NEB chemically component cell transformation with 1 ul of pFC9VR14 (mini prep) and VR22C7 and C4 mini preps (both aligned with VR22 reference when previously sequenced) using homemade chemically competent cells. After 1 hour recovery at 37C, plated 250 ul (1/4th total volume) onto AMP supplemented plates and placed in 37C room at 3:30 PM to grow overnight.
12/22/21 #
12/21/21 midi prep sequencing: Reaction setup #
Sending midi preps completed yesterday for sequencing. Prepared samples with 3 ul of each prep. Quintara order information below.
| Label | Name | Type | Size(kb) | Conc(ng/ul) | Premixed | Name | Conc(uM) | Special Handling |
|---|---|---|---|---|---|---|---|---|
| 1 | pFC9VR4-midi | Plasmid | 3.5 | 80 | Yes | pFC9_t7_primer_1 | 1.67 | GC rich |
| 2 | pFC10VR7-midi | Plasmid | 3.5 | 80 | Yes | pFC9_t7_primer_1 | 1.67 | GC rich |
| 3 | pFC11VR8-midi | Plasmid | 3.5 | 80 | Yes | pFC9_t7_primer_1 | 1.67 | GC rich |
| 4 | pFC12VR14-midi | Plasmid | 3.5 | 80 | Yes | pFC9_t7_primer_1 | 1.67 | GC rich |
| 5 | pFC13VR15-midi | Plasmid | 3.5 | 80 | Yes | pFC9_t7_primer_1 | 1.67 | GC rich |
| 6 | pFC14VR16-midi | Plasmid | 3.5 | 80 | Yes | pFC9_t7_primer_1 | 1.67 | GC rich |
| 7 | pFC15VR17-midi | Plasmid | 3.5 | 80 | Yes | pFC9_t7_primer_1 | 1.67 | GC rich |
| 8 | pFC16VR18-midi | Plasmid | 3.5 | 80 | Yes | pFC9_t7_primer_1 | 1.67 | GC rich |
| 9 | pFC17VR22-midi | Plasmid | 3.5 | 80 | Yes | pFC9_t7_primer_1 | 1.67 | GC rich |
| 10 | pFC18VR24-midi | Plasmid | 3.5 | 80 | Yes | pFC9_t7_primer_1 | 1.67 | GC rich |
| 11 | pFC19VR31-midi | Plasmid | 3.5 | 80 | Yes | pFC9_t7_primer_1 | 1.67 | GC rich |
Midi prep VR22 C4, C7 and VR14: Preparation #
Set up cultures (150 ml LB 100 ug / ml AMP) inoculated with single colony and placed on second floor shakers 37C 200 rpm at 3 PM.
12/23/21 #
Midi prep VR22 C4, C7 and VR14: Execution #
Ran through midi prep protocol for colonies transformed using VR22C4 and C7 minipreps. Both of these preps where previously sequenced and aligned with VR22 reference with high confidence. Also included four VR14 samples since previous prep gel results showed high amounts of dimer. Completed Isopropanol precipitation at 8:50 AM. Started preps at 7 AM.
Agarose gel #
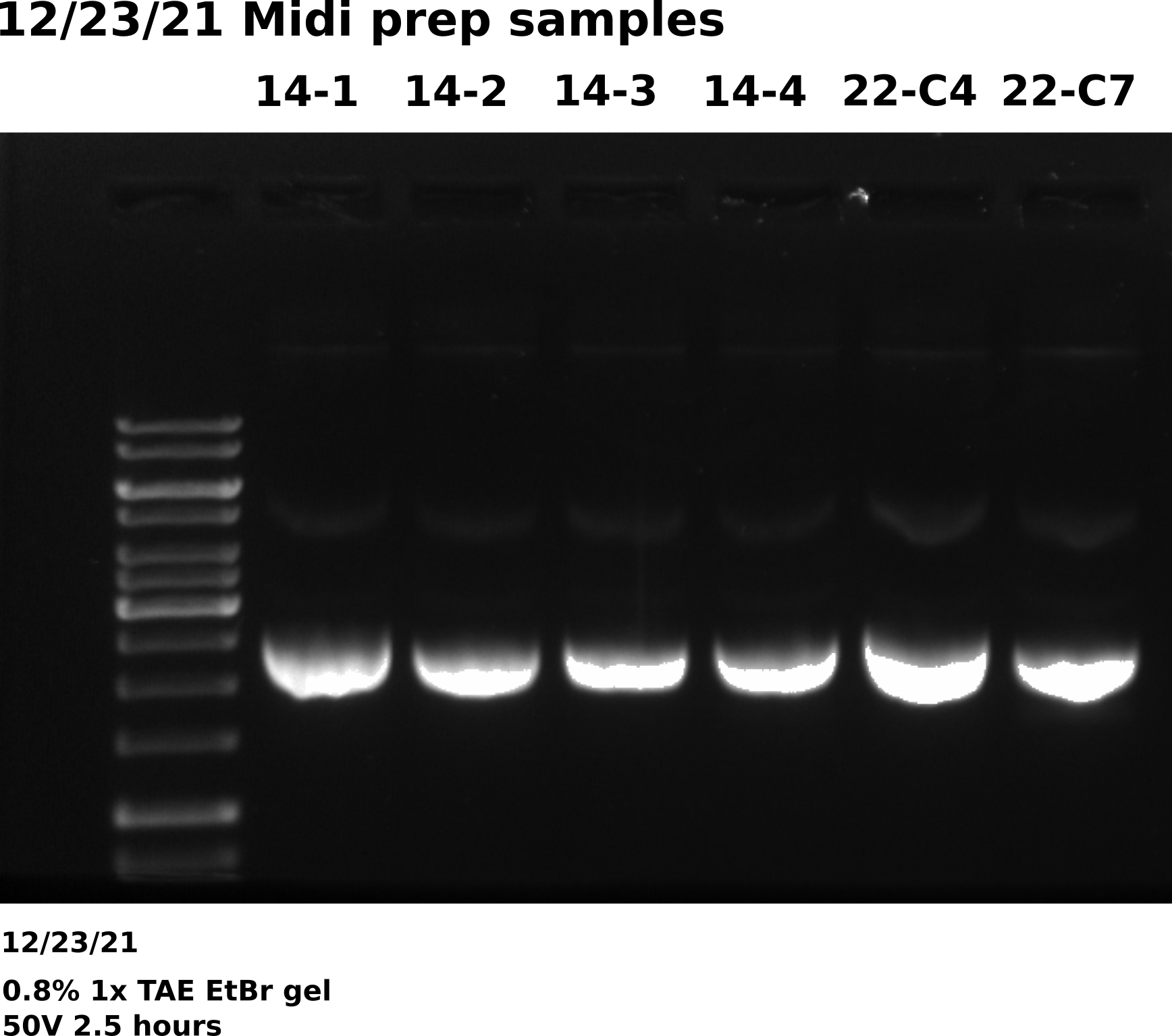
Overall gel looks good, bands are at the correct height with little to no dimer in any of the samples. Will send for sequencing when return from break.
12/21/21 midi prep sequencing: Analysis #
Added runs to midi prep lane of the VR-verify pipeline and reran.
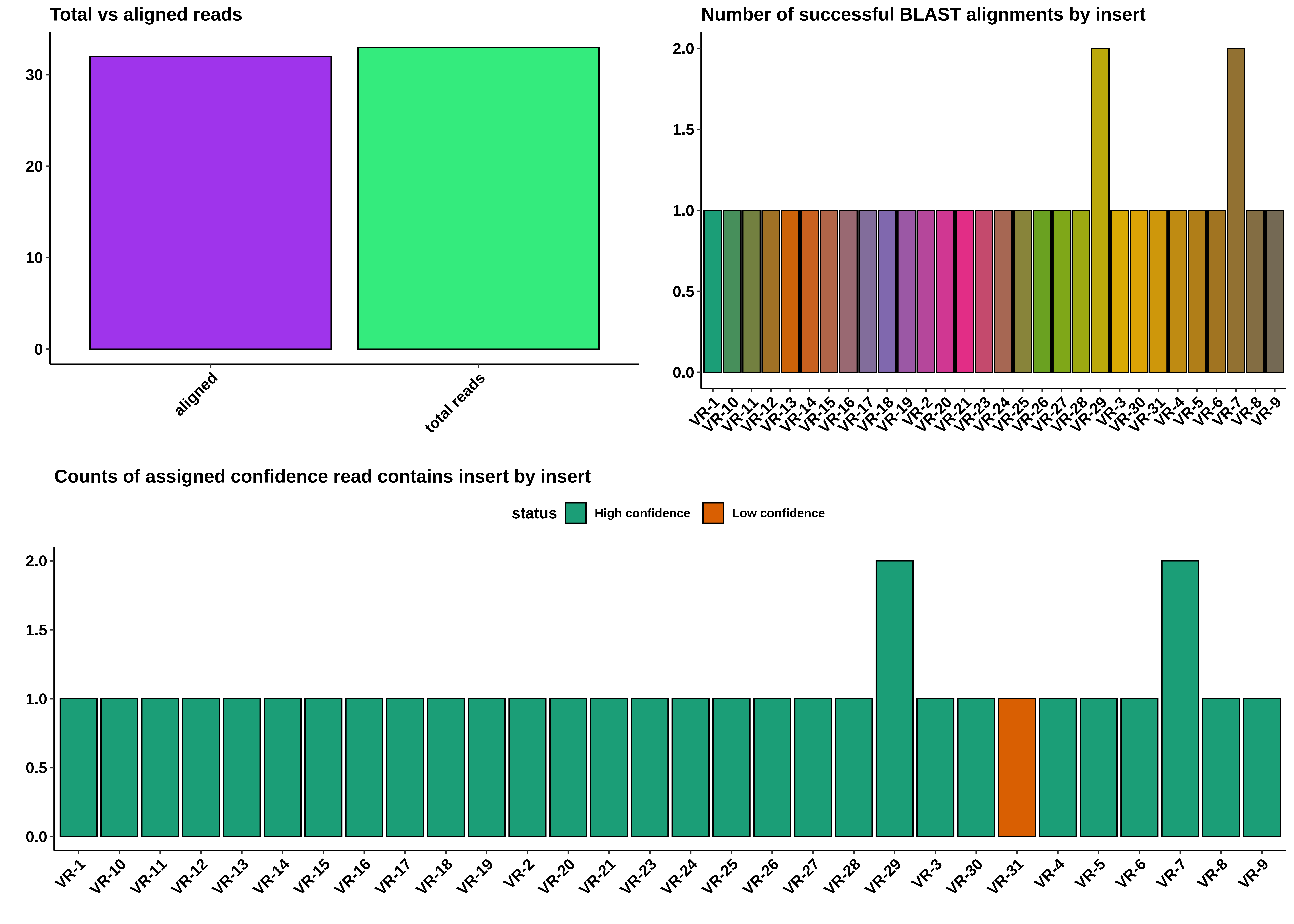
Overall results look pretty good. Most new sequences have high confidence alignments, save VR31. Additionally VR7 can sort of be ignored here because the agarose gel showed that sample to be mostly dimer.
VR31 detail #
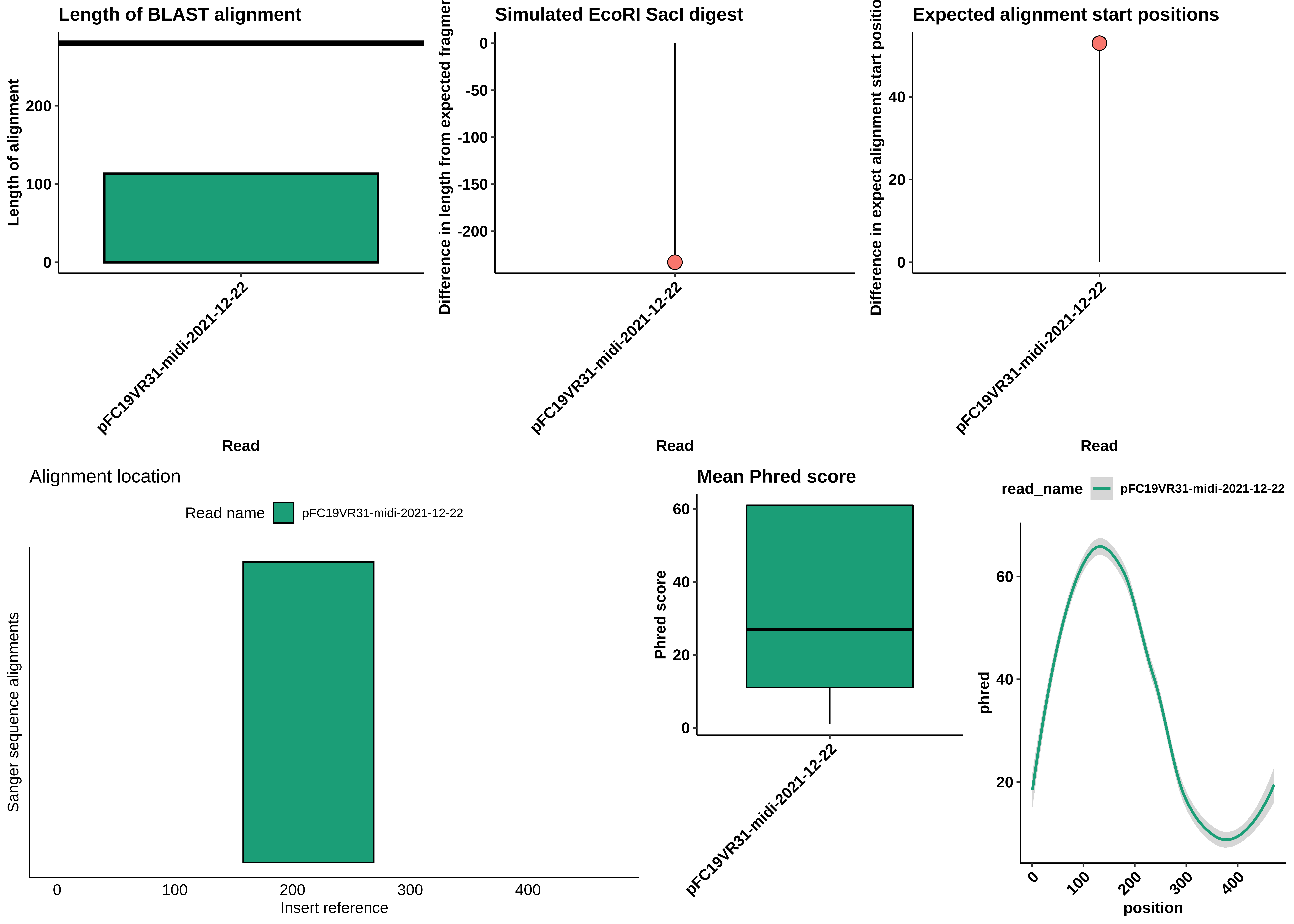
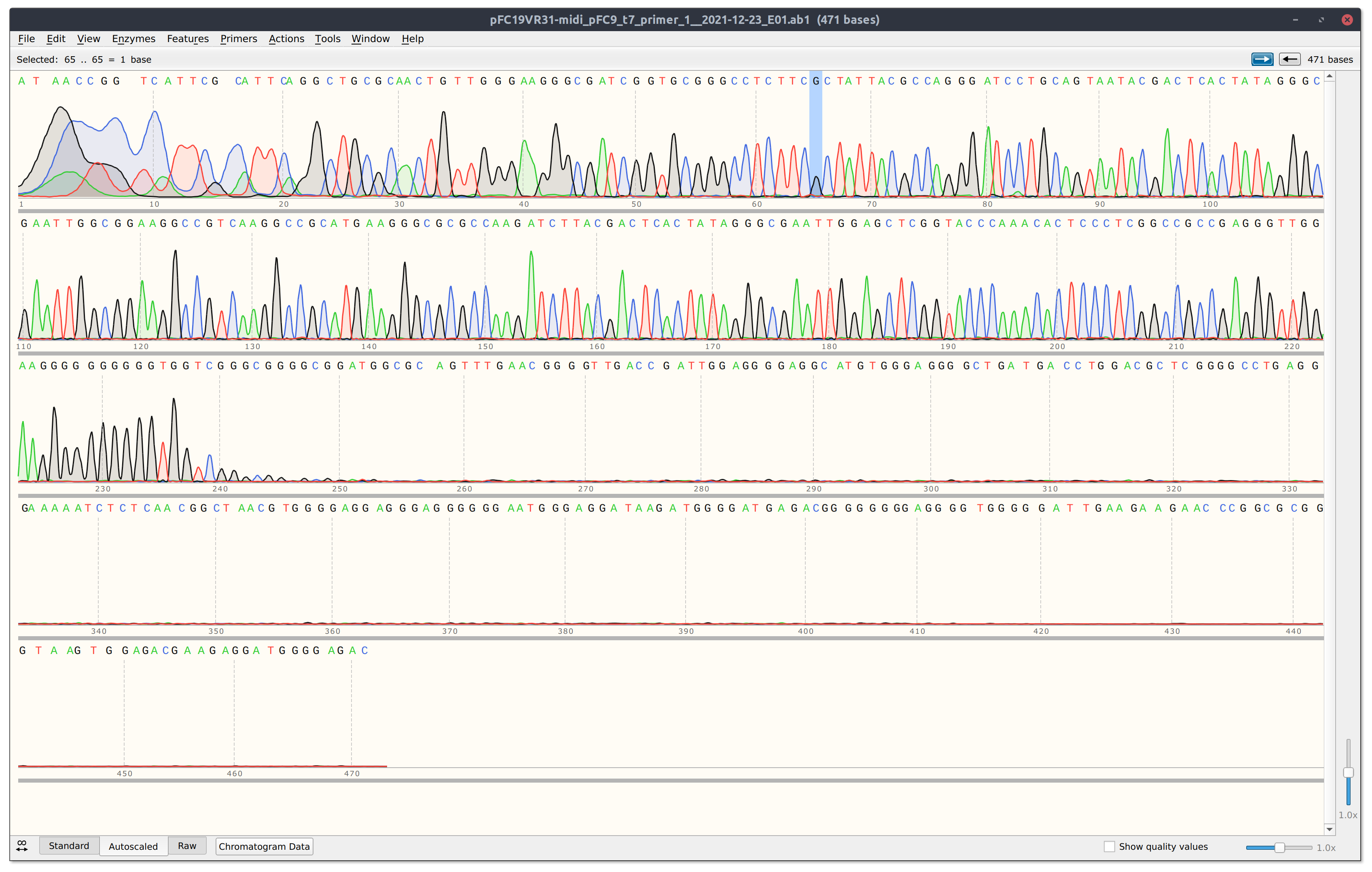
Trace is strong but read is short, likely due to the reaction dying at G rich stretch of the insert. When I come back from break will sequence in the reverse direction.
1/5/21 #
VR14 midi preps #
I had previously written in my notes that the VR7 midi prep needed to be redone because of visible amount of dimer in the gel (see 12/20/21 midi prep gel). However, this was incorrect had has been corrected, it was actually VR14 that showed high dimer. While I continued to write VR7 in my notes, when I re-did the preps on 12/23 I used VR14. However, over winter break looking back at my notes to figure out what I needed to do once I was back in Davis I saw VR7 and forgot I had actually used VR14 and had Tada’s set up VR14 cultures yesterday, thinking VR14 preps were still incomplete. I midi prepped those cultures earlier this morning before realizing this mistake. I prepped 4 VR14 samples following standard lab protocol and re-suspended in 1 ml Tris HCl after isopropanol precipitation. Then used 0.5 ul each sample for agarose gel to check if dimer was present. While gel was running digested samples with RnaseA for 45 minutes and then added 500 ul LiCl and put all samples into freezer.
Agarose gel check #
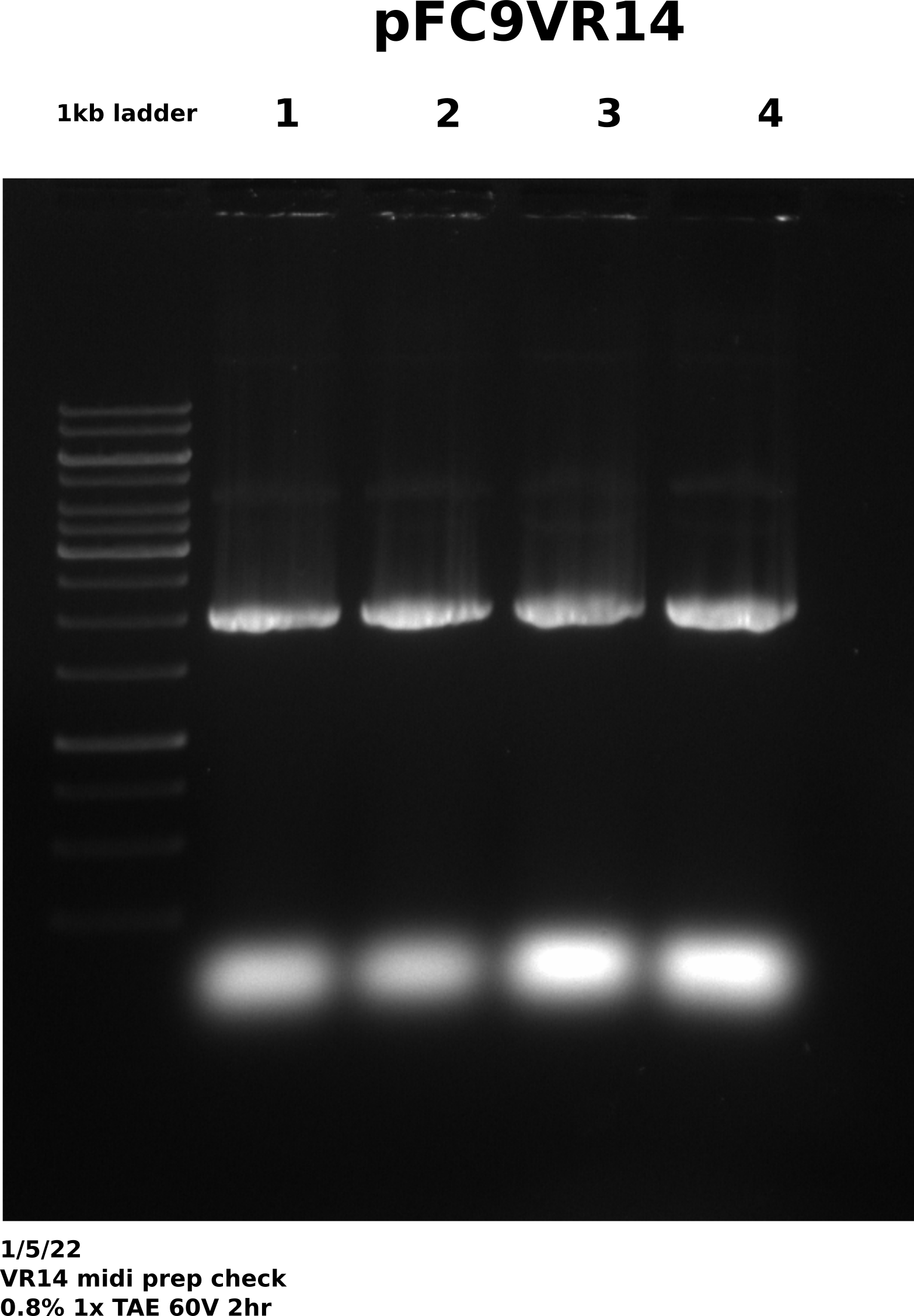
All samples look good with minimal amount of dimer present. If VR14 samples from 12/23 do not look good after sequencing will then sequence these as a backup.
T7 from Fraiser lab #
Fred picked up 1 ml T7 made by Fraser lab. Having Tada’s test by IVT with pFC9 as the template and serial dilutions of the T7 sample.
1/6/21 #
Midi prep sequencing results #
VR14 #
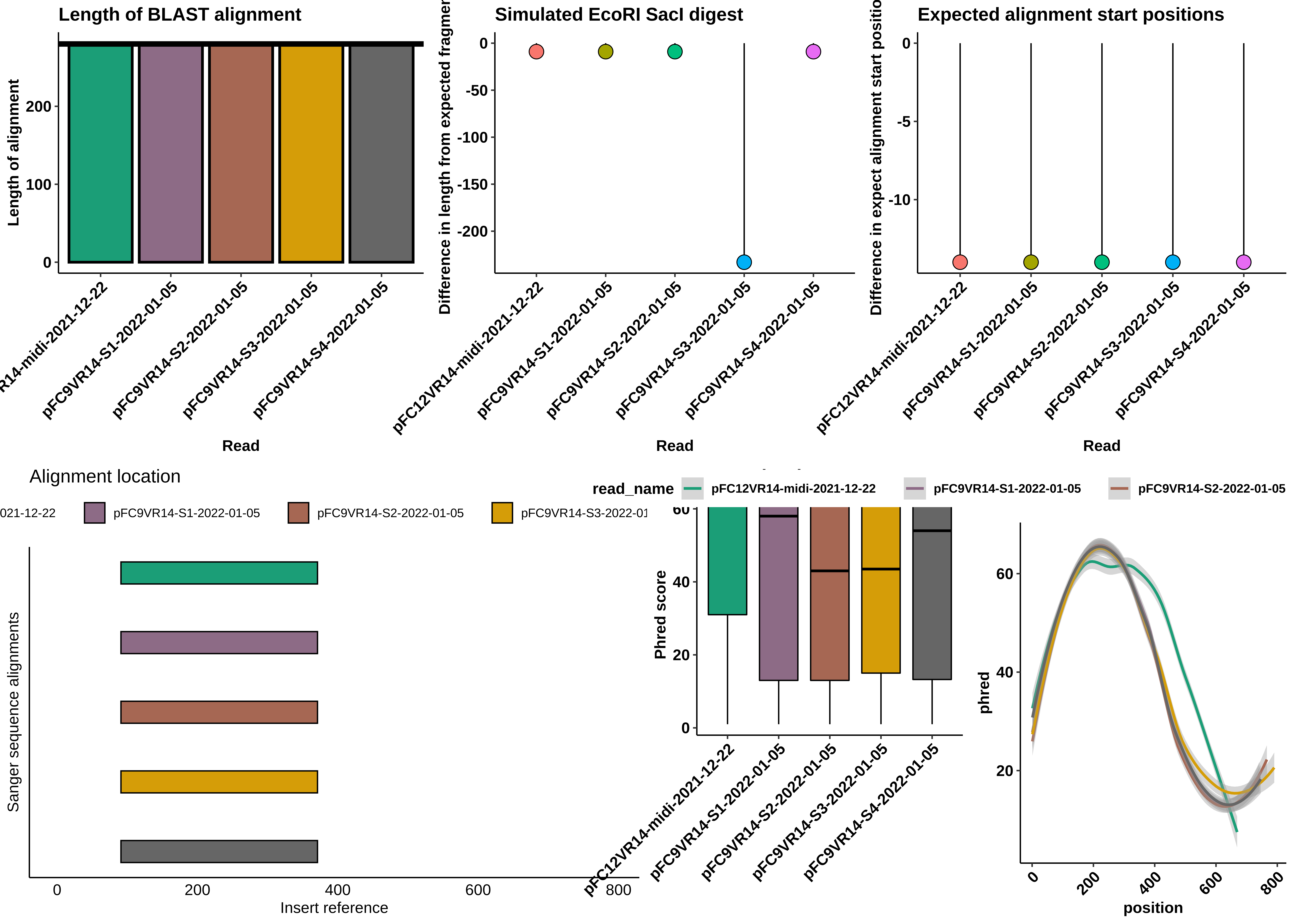
Sent four VR14 samples in for sequencing which were mostly monomer based on agarose gel results. Location of and strength of alignments looks good for all of them. EcoRI-SacI sim digest is off for pFC9VR14-S3, but expected alignment start is correct. Maybe error in reading a base at the digest site. Worth looking closer at if going to be used but for now will use pFC9VR14-S1 moving forward.
VR22 #
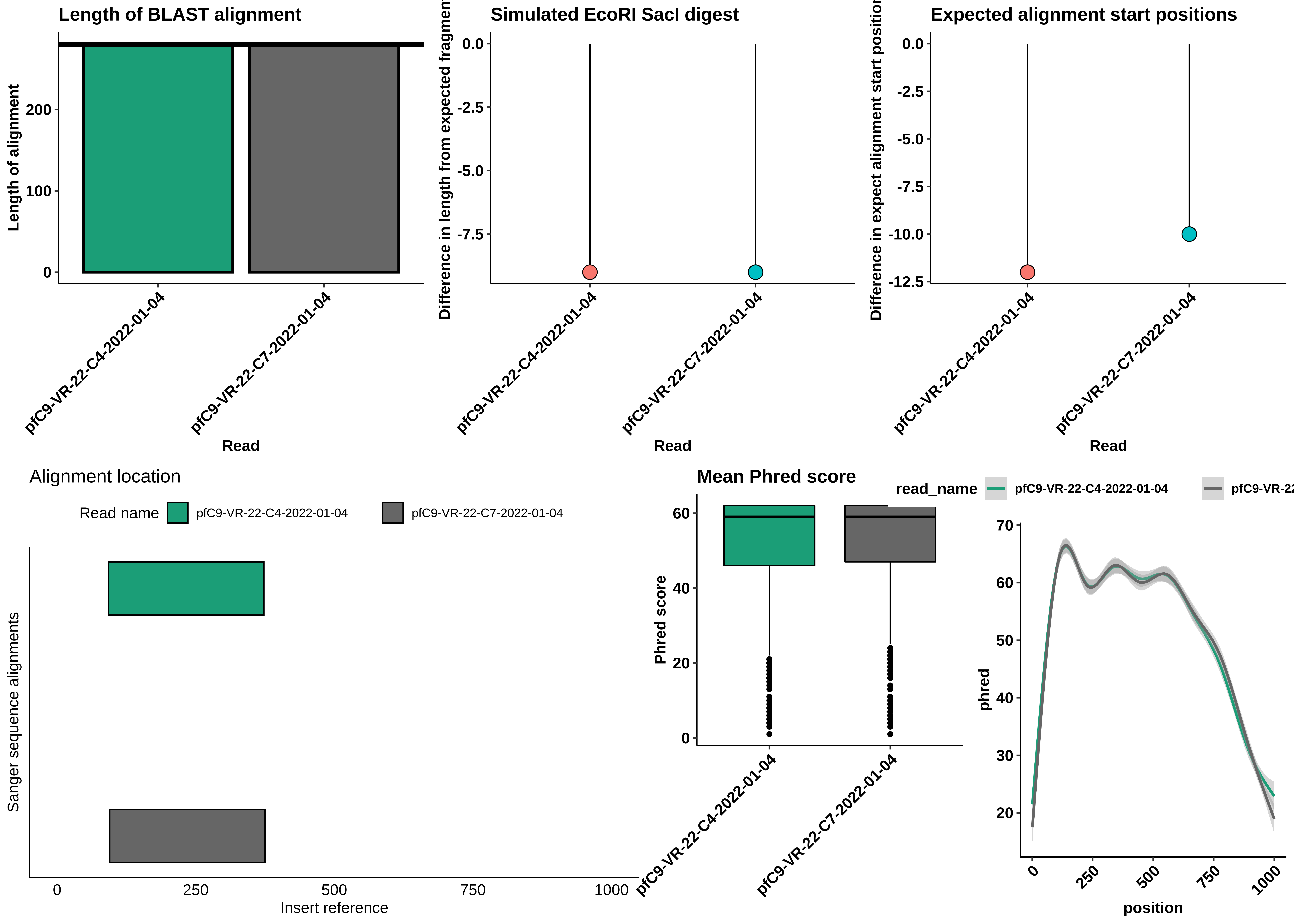
Both VR22 samples sent in look acceptable. Simulated digests and start positions are slighly off but nothing dramatic. Traces and phred scores also strong. Will use C4 sample moving forward.
VR31 #
This is the second time around sequencing VR31 since
first read died in GC rich region. This time I sequenced using pFC9 T7 primer 2 but forgot that
the region this primer binds to is removed in the insertion of the VR insert region so new results were also uninformative. Will need to re-sequence on Friday.
Dialysis test #
Almost all samples showed some level of RNA contamination when visualized via agarose gel. The RNA appears to be mostly digested given how low it tends to run so at Fred’s suggestion I am testing using dialysis tubes to remove RNA from samples. Testing with pFC9VR14-2,3,4 from 12/23 midipreps using 12-14k bags in 4L TE at 4C for 3.5 hours with slow stirring. Before adding samples to dialysis bags I reserved 50ul in case anything goes wrong and to compare on a gel after dialysis is completed.
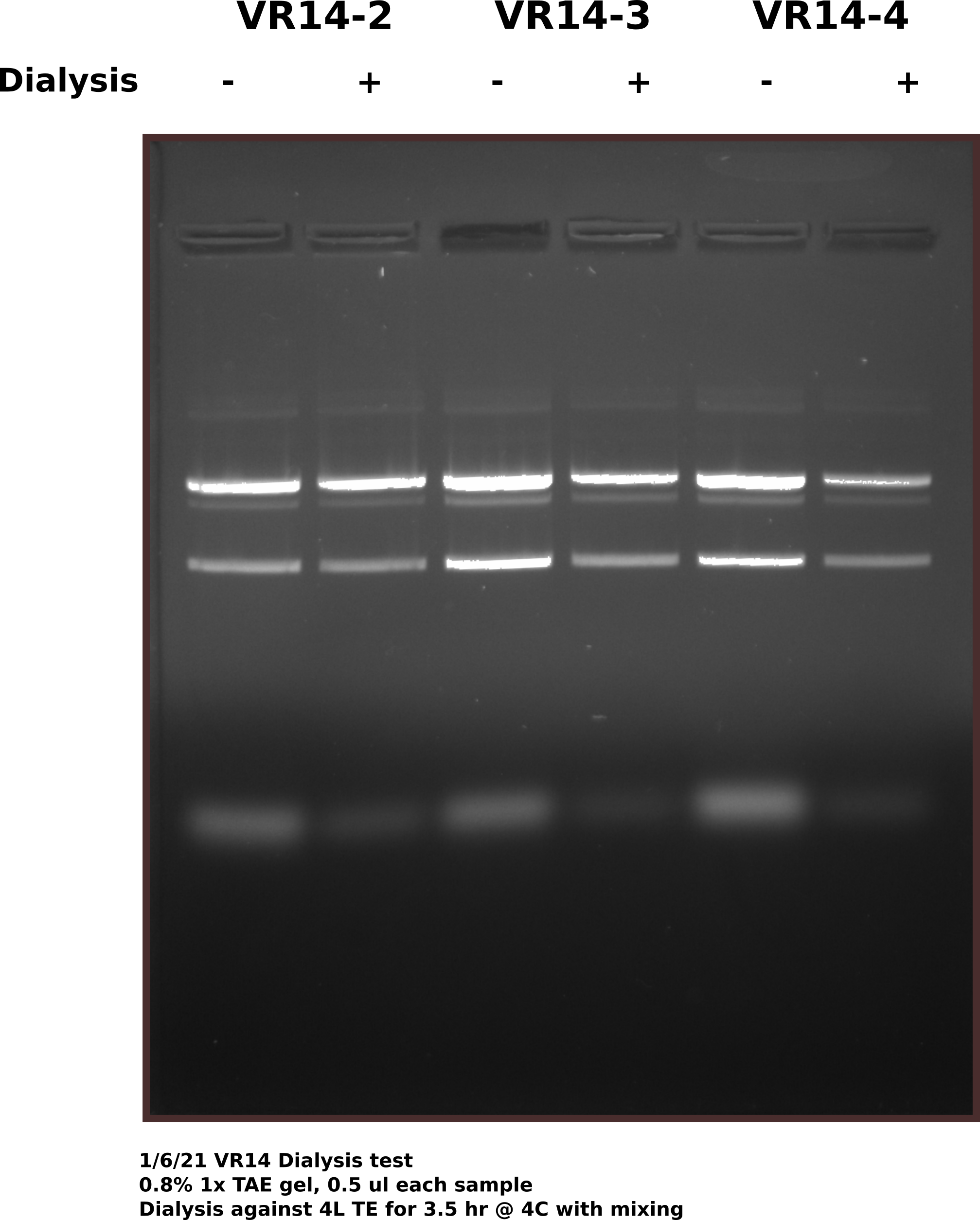
While the dialysis did appear to remove maybe about 2/3 of the RNA from each sample what is more striking and concerning is the amount of top (likely nicked band) that is present in each of the samples. These are the same samples that are shown in the 12/23 midi prep gel and yet there is much more nicked band. This could mean there is a small amount of nickase :( still present in the sample as these plasmids have only gone through one freeze thaw cycle. To test if this is a problem in all samples will run all midi preps out on gel tomorrow and look for increased nick band. Also will add 1 mM EDTA to all samples as a precaution.
1/7/22 #
All midi prep gel (nickase check) #
At time of writing waiting for Tada’s to send me gel images but looks like almost all samples ave increased level of relaxed plasmid compared to when first visualized (fresh after purification from cultures).
VR31 midi prep check #
Send VFC9VR31 midi prep sample for sequencing using VRI primer 2 (reverse primer).
1/10/22 #
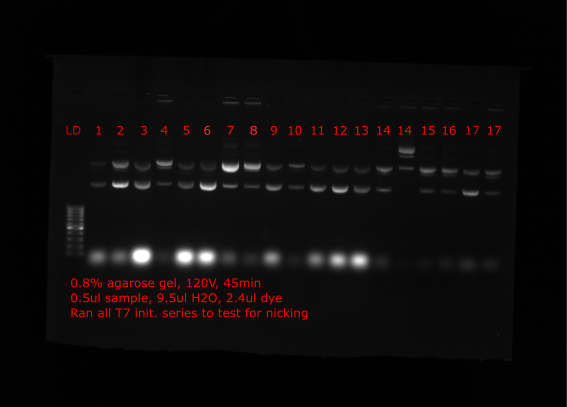
Nick check #
To check if the preped samples seem to be increasing in proportion of relaxed plasmid (assuming nicked) Tadas ran all samples on gel which are shown below. Lanes are labeled with insert number of plasmid.
Top lanes #
Bottom lanes #
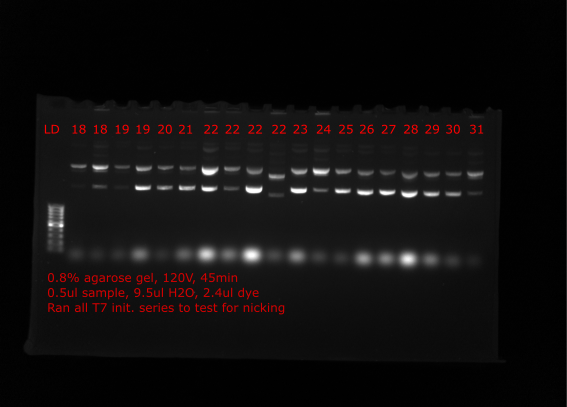
Comparing many of the samples to when run on gels at the time of the preps many samples appear to have much more relaxed plasmid (VR4, VR18 etc.) Solution came up with is to nick all plasmids with nicking endonuclease, then repair nicks with Taq ligase and finally restore supercoiling with DNA gyrase.
Extended dialysis: Setup #
Testing if extending dialysis will remove a greater amount of digested RNA from midi prep samples. Dialyzing in same conditions as 1/6/21 test. Started dialysis at 4 PM.
1/11/22 #
Extended dialysis: Results #
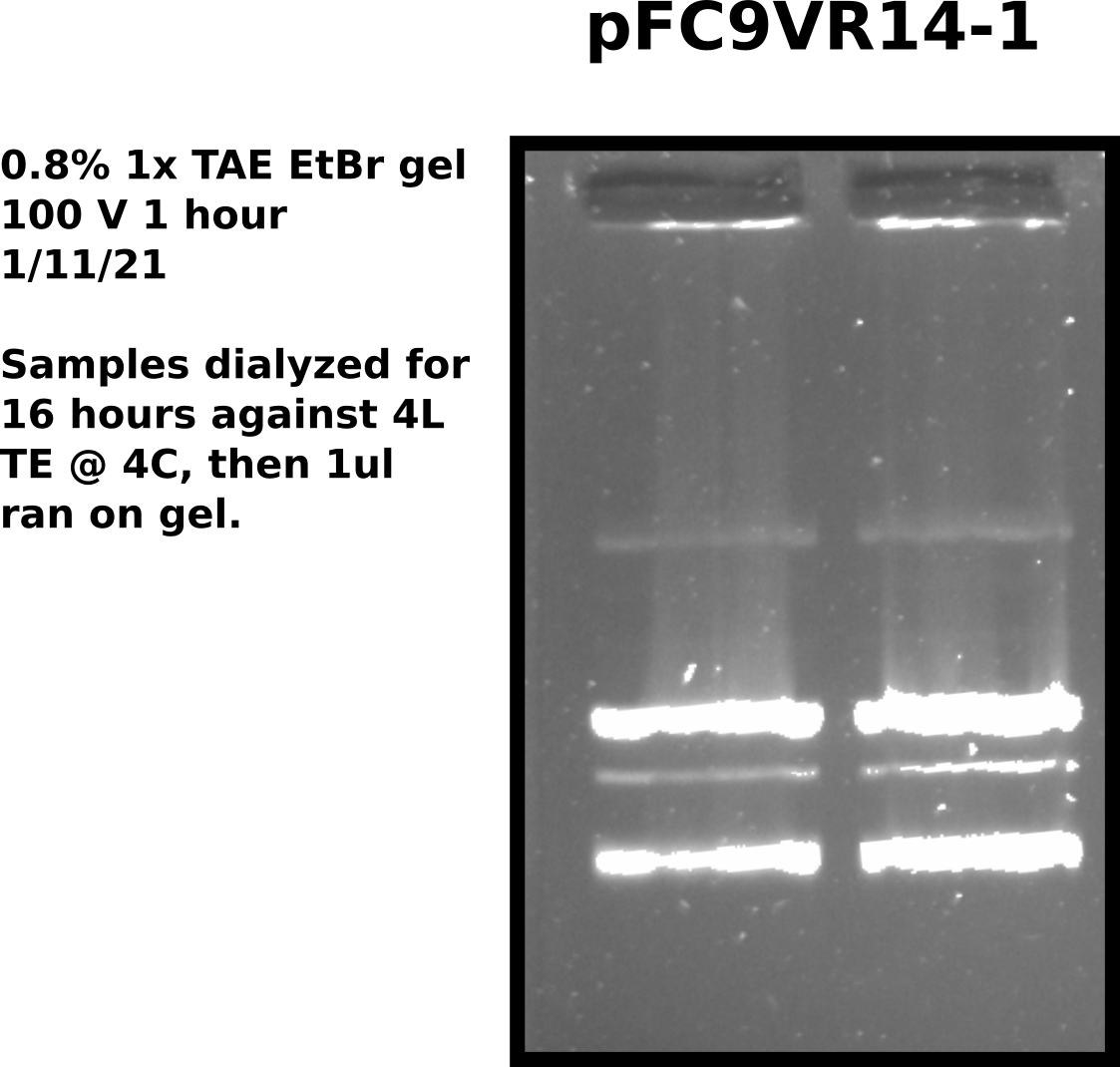
Accordingly, set up dialysis of samples pFC9VR1-8 in same TE buffer to dialyze until 12 PM tomorrow (24 hours). Saved remaining 100 ul in original tubes.
1/12/22 #
Start dialysis for remaining samples #
Started dialysis against TE for remaining samples 9-31 at 10:00 AM. Allow to dialyize for at least 24 hours.
1/13/22 #
Finish dialysis remaining samples #
Removed all samples from TE buffer around 12:00 PM at which point I realized VR15 and 16 samples were not dialyized so they are not represented here. After lab meeting ran all samples out on a gel. Gel is currently not labeled with insert number as some wells were missed during loading (trying to get out of lab) and the goal was to visualize remaining RNA in the samples.

Gel conditions #
Ran at 160V for 30 minutes in 1x TAE buffer with 1 ug / 100 ml EtBr in the gel and buffer. Imaged on second floor imager.
RNA is clearly still present in all samples indicating the dialysis was not successful. Given the small size of the RNA fragments this may indicate that the pores of the tubing have closed up. The tubing is likely 10+ years old at this point. Since this digested RNA is mostly just an aesetic concern at this point will leave samples for now and use agarose gel with ladder to measure concentrations of samples.
1/14/21 #
T7 init midi prep concentration measurements via agarose gel #
In order to measure concentrations of midi preps, running agarose gel of all samples and comparing band intensities against ladder. Complete description of gel layout can be found at this link.
Agarose gel #
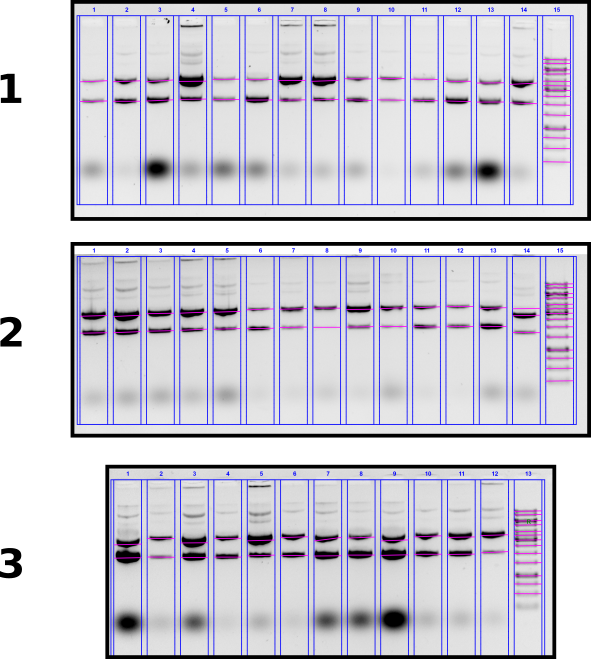
Gel conditions
0.8% agarose 1x TAE buffer 1 ul EtBr / 100 ml buffer in gel and in running buffer. Run for 2 hours at 60V.
Quantifiacation #
I calculated concentrations of midi preps relative to 6000 bp band of 1kb GeneRuler. All calculations done in this Jupyter notebook.
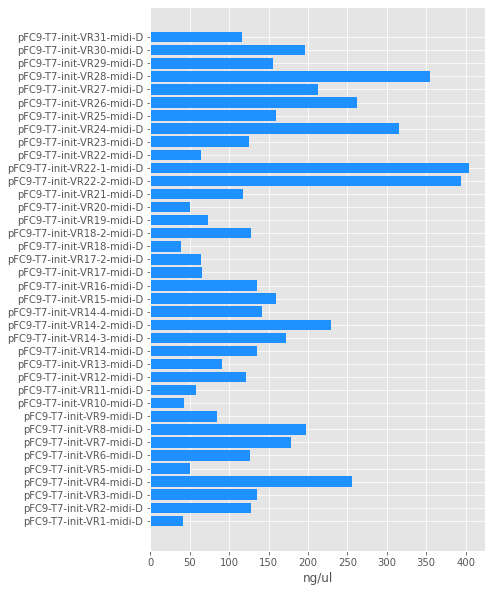
Concentrations ranged from ~50 ng / ul to 400. Sample volumes are not all equal due to dialysis. Quantifications for all samples shown in the table below.
| Tube full name | Tube date | nanograms / ul |
|---|---|---|
| pFC9-T7-init-VR1-midi-D | 12/18/21 | 40.886663999999996 |
| pFC9-T7-init-VR2-midi-D | 12/18/21 | 127.29788400000001 |
| pFC9-T7-init-VR3-midi-D | 12/14/21 | 135.733584 |
| pFC9-T7-init-VR4-midi-D | 12/21/21 | 255.807468 |
| pFC9-T7-init-VR5-midi-D | 12/14/21 | 50.113952 |
| pFC9-T7-init-VR6-midi-D | 12/14/21 | 125.991516 |
| pFC9-T7-init-VR7-midi-D | 12/21/21 | 178.63568800000002 |
| pFC9-T7-init-VR8-midi-D | 12/21/21 | 197.337952 |
| pFC9-T7-init-VR9-midi-D | 12/14/21 | 84.075264 |
| pFC9-T7-init-VR10-midi-D | 12/14/21 | 43.093904 |
| pFC9-T7-init-VR11-midi-D | 12/14/21 | 57.284528 |
| pFC9-T7-init-VR12-midi-D | 12/14/21 | 120.685796 |
| pFC9-T7-init-VR13-midi-D | 12/14/21 | 91.114744 |
| pFC9-T7-init-VR14-midi-D | 12/23/21 | 135.38392 |
| pFC9-T7-init-VR14-3-midi-D | 12/23/21 | 172.40574400000003 |
| pFC9-T7-init-VR14-2-midi-D | 12/23/21 | 228.817484 |
| pFC9-T7-init-VR14-4-midi-D | 12/23/21 | 141.92878 |
| pFC9-T7-init-VR15-midi-D | 12/21/21 | 159.393248 |
| pFC9-T7-init-VR16-midi-D | 12/21/21 | 135.422504 |
| pFC9-T7-init-VR17-midi-D | 12/12/21 | 65.152808 |
| pFC9-T7-init-VR17-2-midi-D | 12/21/21 | 64.474648 |
| pFC9-T7-init-VR18-midi-D | 38.319708 | |
| pFC9-T7-init-VR18-2-midi-D | 12/21/21 | 127.196664 |
| pFC9-T7-init-VR19-midi-D | 12/5/21 | 59.78265999999999 |
| pFC9-T7-init-VR19-midi-D | 12/12/21 | 73.107804 |
| pFC9-T7-init-VR20-midi-D | 12/12/21 | 50.046388 |
| pFC9-T7-init-VR21-midi-D | 12/12/12 | 117.786284 |
| pFC9-T7-init-VR22-2-midi-D | 12/21/21 | 98.68143599999999 |
| pFC9-T7-init-VR22-1-midi-D | 12/12/21 | 404.613748 |
| pFC9-T7-init-VR22-midi-D | 12/23/21 | 63.746984 |
| pFC9-T7-init-VR22-2-midi-D | 12/23/21 | 394.525152 |
| pFC9-T7-init-VR23-midi-D | 12/12/21 | 124.53795200000002 |
| pFC9-T7-init-VR24-midi-D | 12/21/21 | 315.858676 |
| pFC9-T7-init-VR25-midi-D | 12/12/21 | 159.032328 |
| pFC9-T7-init-VR26-midi-D | 12/12/21 | 262.635128 |
| pFC9-T7-init-VR27-midi-D | 12/12/21 | 213.09761200000003 |
| pFC9-T7-init-VR28-midi-D | 12/12/21 | 355.16398399999997 |
| pFC9-T7-init-VR29-midi-D | 12/12/21 | 155.904812 |
| pFC9-T7-init-VR30-midi-D | 12/12/21 | 196.0798 |
| pFC9-T7-init-VR31-midi-D | 12/21/21 | 116.571588 |
Can also be downloaded with wget https://raw.githubusercontent.com/EthanHolleman/misc-jupyter-notebooks/main/T7-init-concentrations/t7InitSeriesConcentrationQuants.tsv.