Background #
In order to remove superhelicity from analysis of sequencing results nicked templates will be linearized before transcription. I am currently planning on using BamHI-HF for T7Init series plasmids as it is a single cutter just upstream of the T7 promoter.
Regardless of the enzyme used, when linear substrates are seen on the gel, no shifting will be observed even with transcribed samples as there is no supercoiling to relax in the first place. It therefore becomes necessary to “super-shift” bands by incubating with S9.6 antibody.
Robert’s paper used this technique to show R-loop formation over linearized substrates, however the supp materials are not very detailed with the protocol used and don’t say much more than “incubate with S9.6 antibody”
“To visualize potential R-loops in relaxed substrates for which further DNA relaxation can’t be detected (e.g. relaxed, nicked and linear substrates), the products were purified by phenol/chloroform extraction and ethanol precipitation, resuspended in TE and incubated with the S9.6 antibody to supershift any RNA:DNA hybrids.” – Stoltz et al.
So I turned to the DRIP protocol to get more specific parameters. Link to complete protocol. I am really only interesting in S9.6 antibody incubation part; however I am not doing any pull down on samples since I want to see DNA that was not bound by S9.6. Relevant section of the protocol is screenshotted below.

Think will need to greatly reduce the volume as the amount of DNA I would be using will be much lower. The only downstream analysis that will be happening with these samples will be agarose gel. I am currently planning on just scaling this protocol down linearly. Generally, I use around 200 ng of DNA in an IVT reaction so somewhere around that scale for DNA. Concentration needs to be high enough so that can run 10-20 ul of sample on the gel. The volume that sample is incubated with S9.6 with will be the final volume since cannot do any steps that would de-proteinize the sample.
Table below shows a possible S9.6 incubation setup for a 100 ul total reaction volume. All volumes in ul, all masses in ng.
| Total Volume | DNA Volume | DNA concentration | 10x Binding Buffer | S9.6 | TE | Final DNA concentration | Volume Sample for gel |
|---|---|---|---|---|---|---|---|
| 100 | 20 | 10 | 10 | 2 | 68 | 2 | 25 |
Running 25 ul of S9.6 incubated sample should plenty to see on a gel.
Testing S9.6 supershifting #
3/15/22 #
Restriction digests #
Digested 5 ug of T7Init Mix 0 with BamHI-HF and another 5 ug with BsaI-HF. While BamHI is my prefered enzyme for future reactions due to its location just upstream of target site I was worried that its extreme proximity may cause issues with transcription which is why I also included the BsaI-HF digest whos cut site is much further away from T7 + 1 site.
Total reaction volume was 150 ul.
In vitro transcrition #
After digesting samples for 2 hours at 37C placed on ice. Then removed 600 ng of each sample (18.01 ul of each digest) and set up T7 IVT experiment according to the lab protocol. Did not include RnaseH control, only untranscribed.
After incubating at 37C for 20 mins in thermocycler removed and added EDTA to final concentration of 36 mM and placed on ice.
Incubation with S9.6 antibody #
Prepared S9.6 incubations with transcribed and un-transcribed samples. Transcribed sample reactions were prepared as described by the table below (all units are in ul).
| Total Volume | DNA Volume | DNA concentration | 10x Binding Buffer | S9.6 | TE | Final DNA concentration | Volume Sample for gel |
|---|---|---|---|---|---|---|---|
| 100 | 20 | 10 | 10 | 2 | 68 | 2 | 25 |
Since total volume of untranscribed reactions was only 9.66 (following lab protocol) I added 10.34 ul TE to replace the missing volume compared to transcribed samples. All reactions setup on ice. After completing reaction setup placed samples in deli fridge on tube rotator to mix overnight. Incubation began at 4:17 PM.
3/16/22 #
Agarose gel of S9.6 incubated samples #
Removed samples from rotator at 9:00 AM for total incubation time of 17 hours. Aliquots volumes containing 50 ng of DNA from each sample. Transcribed samples contain 2x DNA untranscribed so diluted aliquots of transcribed sample with TE. Added purple loading dye to 1x concentration and ran samples on 1x TBE gel without EtBr at 60V for 2 hours.
Agarose gel image #
No shift was observed.
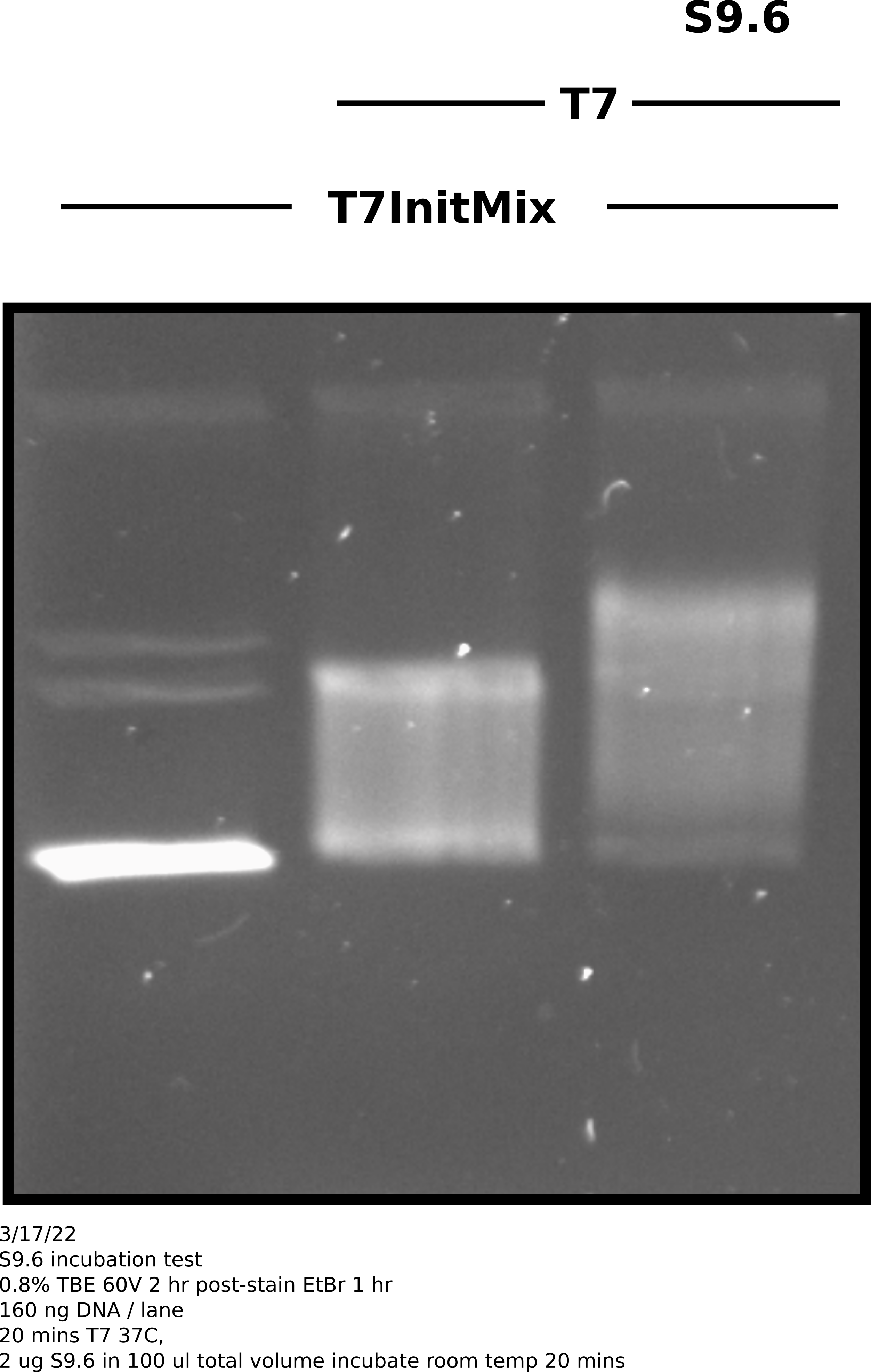
3/17/22 #
Repeated S9.6 treatment conditions from yesterday but instead of using linearized template in the transcription reaction I used uncut T7Init Mix 0 and transcribed according to lab IVT protocol.
S9.6 incubation was preformed at room temperature for 20 mins at Fred’s advice in the same conditions as yesterday (see this table.)
Agarose gel image #
Analysis #
Transcribed plasmids were for sure shifted more when treated by S9.6 but
not nearly as dramatically as is seen in papers that use this same method (
see Stoltz et al
). Also in the future should also have an un-transcribed S9.6 treatment
to show that anitbody is not shifting un-transcribed substrate.
3/21/22 #
Insights from the boss #
After talking with Fred about S9.6 shifting in relation to other Cas9 experiments he remembered that DTT which has a high concentration in transcription reactions (10 mM final) resulting in this case in 5 mM final in the S9.6 binding reaction can destroy S9.6 due to the resolution of di-sulfide bridges and therefore resulting in much weaker shifts.
The solution to this being to preform EtOH precipitation after transcription reactions and then resuspend in small volume of Tris-HCl before continuing on with S9.6 binding.
Effect of DTT on S9.6 binding in in vitro transcription reactions #
Sample prep and reaction protocols #
Prepared pFC9 template according to lab in vitro transcription protocol. Left one sample (200 ng) untranscribed and transcribed aliquote containing 400 ng of template. Split the transcribed template into EtOH precipitated and unprecipitated samples. Preformed EtOH precipitation and resuspended sample in 10 ul Tris-HCl.
Increased all sample volumes to 100 ul using 10X DRIP binding buffer, npH20 and 2 ul (2 ug) of S9.6. Incubated all samples at room temperature for 20 minutes before loading 60 ul of each sample onto a TBE agarose gel. Ran samples at 60V for 2 hours before post-staining with EtBr and imaging.
Agarose gel image #

Analysis #
It is unexpected that EtOH precipitated sample looks basically the same as the un-precipitated sample. I realized after viewing that I should have done an RnaseA digestion as it is very possible I am just seeing dsRNA signal which is overcoming any RNA-DNA signal present. Need to repeat with RnaseA digest and probably should double reaction size so more DNA can be included.
3/22/22 #
pFC9 IVT with linerization and S9.6 treatment #
Protocol #
Digested 1800 ng pFC9 with 2 ul BamHI-HF in rCutSmart buffer in total volume 150 ul. Preformed IVT reaction (at 3x size) according to lab protocol with NEB T7 RNA Pol. Stopped reaction with EDTA to final concentration 36 mM. Digsted all samples with 3 ul 1:1000 dilution RnaseA for 10 mins at room temp. Split transcribed samples into EtOH precipitated and no precipitation treatments (30 ul each sample). After EtOH precipitation treated all samples with 2 ug S9.6 antibody in 100 ul 1x DRIP binding buffer diluted with TE. Incubated samples with S9.6 for 20 minutes at room temp before adding 16.66 ul NEB purple loading dye. Loaded 60 ul of each sample onto TBE agarose gel and ran for 2 hr at 60V before post-staining with EtBr.
Agarose gel image #
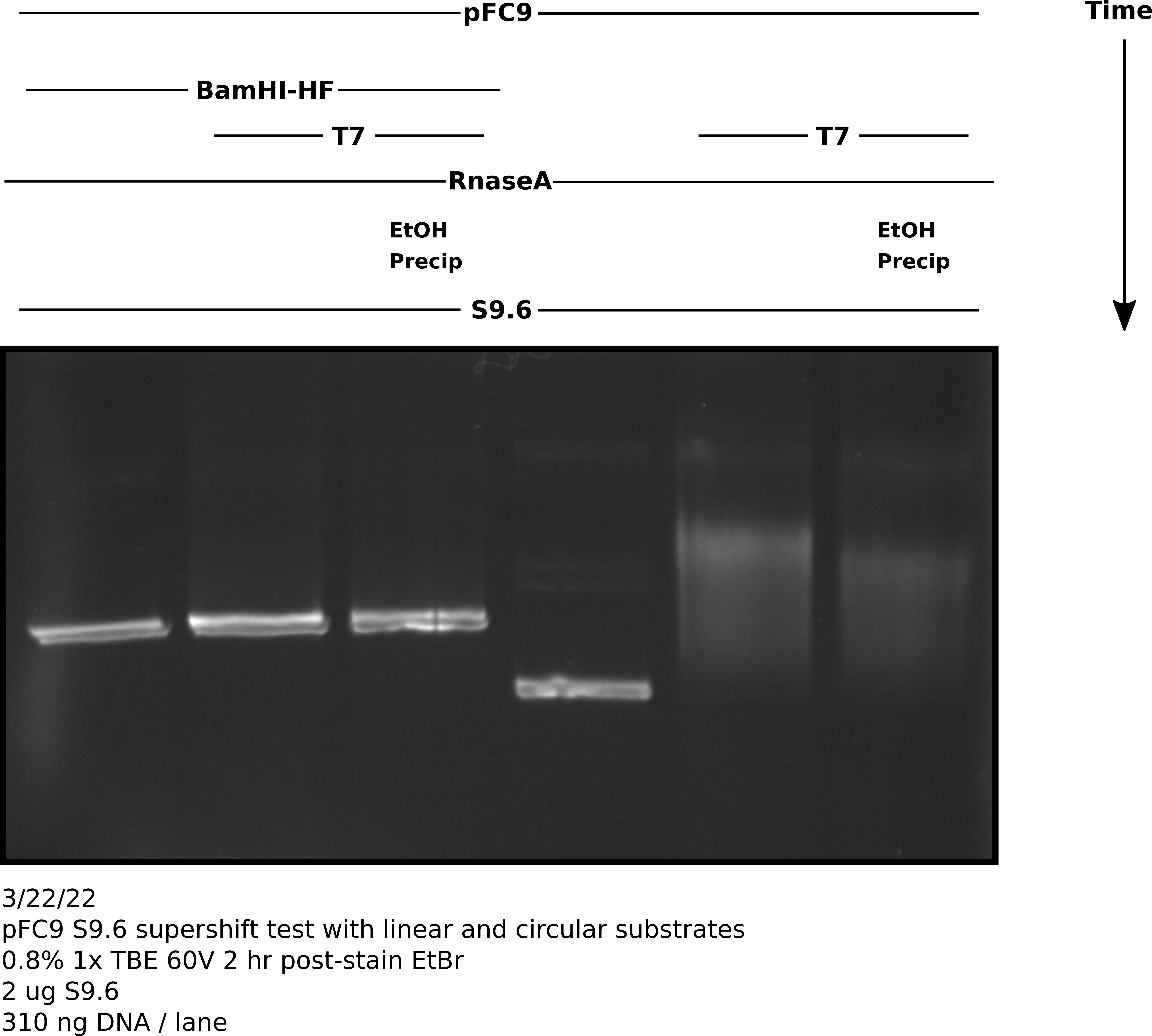
Analysis #
There seems to be about the same amount of shifting that I observed in my previous un-precipitated gel. This got me thinking about what else could be interfering with antibody binding. I realized that the NEB purple loading dye contains a fair amount of SDS even in the 1x concentration.

Quick google on the topic brings up Qualtiere et al, 1977 which states in
the abstract…
Generally, at concentrations greater that 0.01% SDS, almost all immunochemical
reactivity is destroyed.
While antibodies should already be bound by the time the loading dye hits them 0.08% might be enough to denature the vast majority of them. Next follow up gel should include RnaseA digestion, EtOH precipitation, linear and circular samples, S9.6 treated and untreated samples and finally all samples should be loaded with NEB SDS free purple loading dye.
3/23/22 #
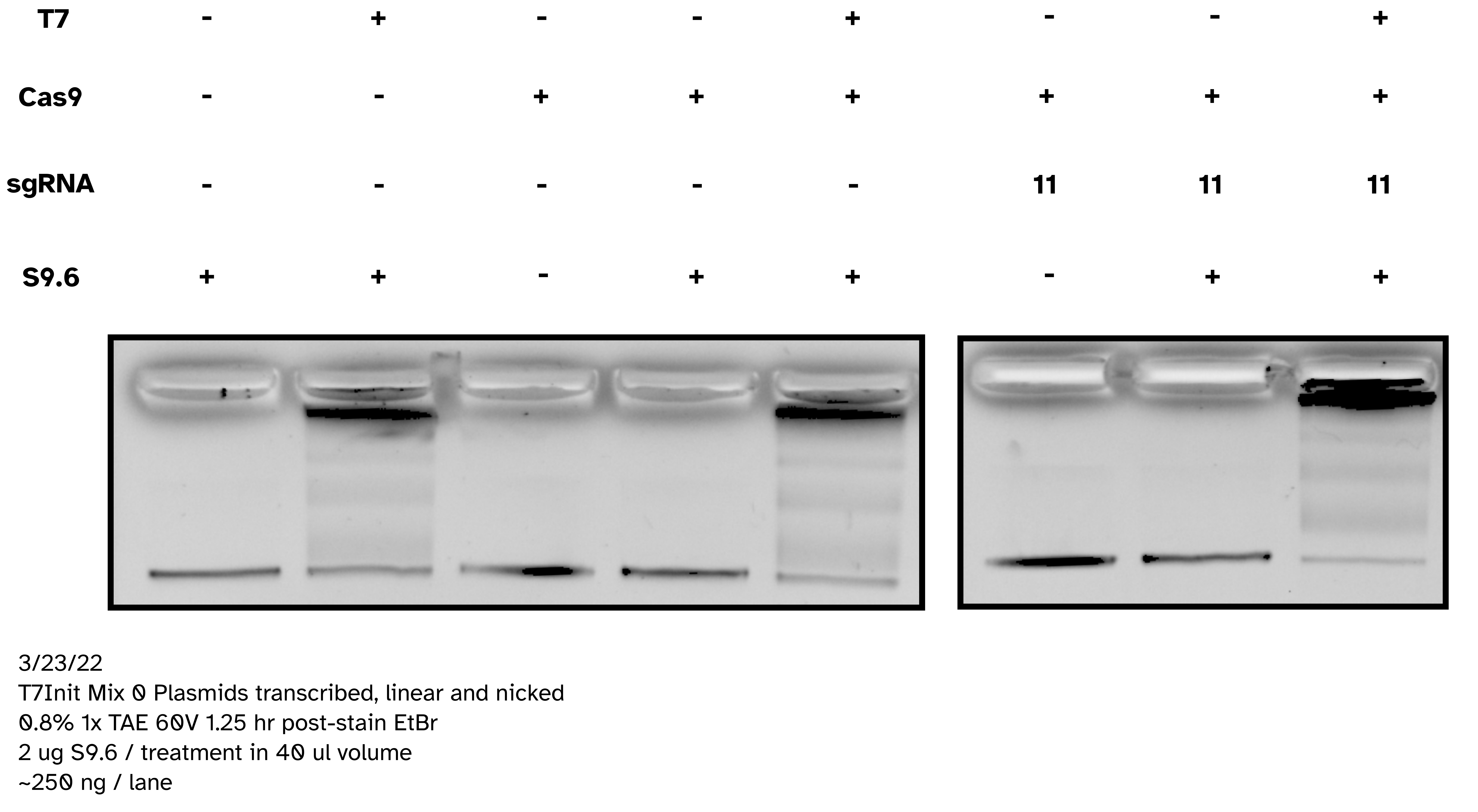
IVT with pFC53 + T3 Pol with and without S9.6 binding; no SDS loading dye #
Today attempting to replicate results from Stoltz et al. paper where
pFC53 was used to demonstrate gel shift upon transcription. Including linearized
and circular transcribed and untranscribed treatments as well as S9.6 treated
and untreated samples. Linearization done using XBaI who’s cut sight is just
upstream of the T3 promoter in pFC53 sequence.
Protocol #
Prepared 4.8 ul pFC53 (2400 ng) with 3 ul NEB 10x rCutSmart buffer and 18.2 ul npH20. Split volume into 13 ul aliquots and added 2 ul XbaI to digested sample and 2 ul npH20 to control. Allowed to digest for 45 mins at 37C before heat inactivating at 65C for 20 minutes. Split each sample again into transcribed and un-transcribed aliquots and preformed IVT according to lab protocol with NEB T3 RNA Pol. After 20 mins at 37C added EDTA to final concentration of 36 mM to stop reactions. Added 2 ul 1:1000 RnaseA and digested for 10 minutes at room temperature before preforming EtOH precipitation on all samples.
Re-suspended samples in 20 ul Tris-HCl and prepared for S9.6 treatment. For each sample added 5 ul precipitated DNA solution, 5 ul 10x DRIP buffer, 38 ul of npH20 and 2 ul 1 mg/ml S9.6 dilution or water depending on if sample was treatment of control.
Samples were then spun down and incubated at room temperature for 20 minutes. After incubation added 8.33 ul 6x Purple loading dye without SDS. Loaded entire sample onto TBE agarose gel and ran for 2 hours at 60V, then post-stained with EtBr.
Agarose gel image #
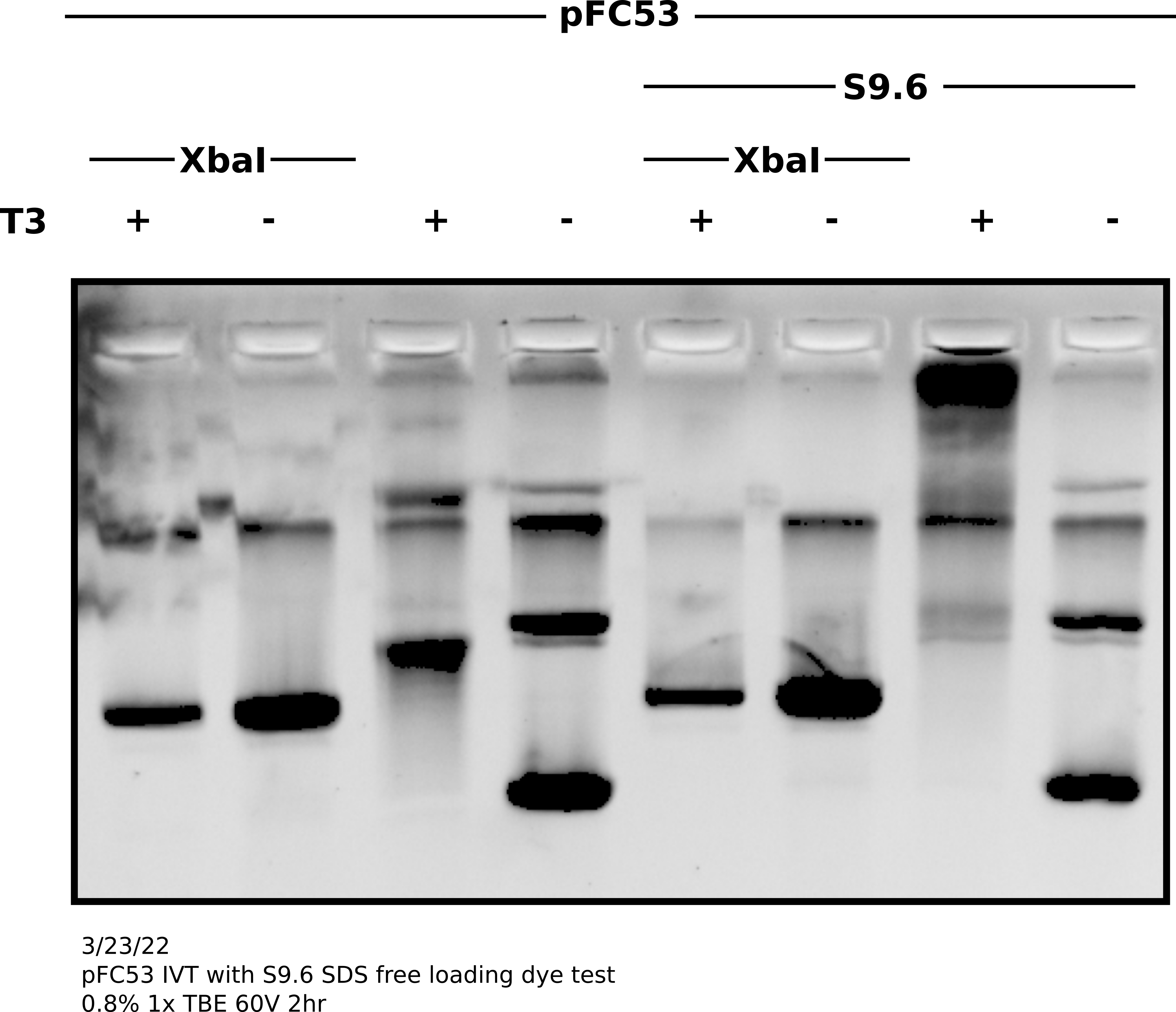
Analysis #
Shift on supercoiled plasmid was good and present without S9.6 indicating IVT reaction was good. Additionally, bit of reduction in S9.6 treated transcribed linear compared to controls indicates binding to linear templates as well. It seems SDS in loading dye was de-naturing antibodies and preventing visualization on agarose gels. In future do not use SDS containing loading dyes for S9.6 supershifted gels.
TODO: Label this gel and stuff #