IVT and bifulfite conversion protocol notes #
IVT protocol #
Looking through lab papers that did IVT at some point did not see specific protocol beyond transcribing DNA for 20 mins, phenol chloroform extract followed by EtOH precipitation and re-suspend in TE. Drawing protocol information from R-Loop Formation Is a Distinctive Characteristic of Unmethylated Human CpG Island Promoters (Ginno et al 2012) and Ultra-deep Coverage Single-molecule R-loop Footprinting Reveals Principles of R-loop Formation. The single molecule footprinting paper (Malig et al 2020) sites Ginno et al 2012 for its IVT protocols.
Bisulfite conversion #
Lab publications use the Zymogen lignting DNA meythlation kit reagents and protocol except for the incubation step. From Malig et al 2020
In brief, the DNA was not denatured before bisulfite
treatment and treatment with the conversion reagent
was performed at 37 C for 2 h with gentle
rotation. Desulfonation and sample recovery were
performed as instructed.
PacBio library prep notes #
UC Davis genome center requirements #
- Minimal DNA purity: OD 260/280 should be 1.8-2.0; OD 260/230 should be >2.0
- Has undergone a minimum of freeze-thaw cycles.
- Has not been exposed to high temps (> 65°C for more than one hour can cause a detectable decrease in sequence quality).
- Has not been exposed to pH extremes (< 6 or > 9).
- Does not contain insoluble material.
- Is RNA-free.
- Has not been exposed to intercalating fluorescent dyes or ultraviolet radiation.
- Does not contain, divalent metal cations (e.g., Mg2+), denaturants (e.g., guanidinium salts, phenol), or detergents (e.g., heme, humic acid, polyphenols). Amplicon samples should be submitted in EB buffer (EDTA-free).
- Must be double-stranded. Single-stranded DNA cannot be converted into SMRTcell templates but can interfere with polymerase binding.
Samples of 10-20kb should contain 20 ug or greater of DNA.
Requirements come from BacPio sample prep recommendations: download link.
Before submitting samples #
- Email picture of samples on agarose gel with marker at least 20 kb upper band
- Access purity via spectrophotometry – the 260/280 ratio should be between 1.8 to 2.0 and the 260/230 ratio should be higher than 2.0.
- Use fluorometric methods (e.g. Qubit) for DNA quantification if possible. Measure each sample at least 3 times and accept only reproducible measurements
Cost #
- 1900 pre SMRT-cell (email ohnguyen@ucdavis.edu)
- 650 for library prep
12/15/21 #
pFC9 IVT and non-denaturing bisulfite treatment #
I wanted to go through the IVT and bisulfite treatment protocol just to go through the motions. So I am testing using pFC9 and plan to evaluate results by directly Sanger sequencing the bisulfite treated samples. This will measure conversions over a population of molecules so in theory when looking at the traces treated with bisulfite I expect to see noisy base calls at conversion sites and clean calls were conversion did not occur.
Protocol #
- Set up reactions according to IVT bisulfite conversion spreadsheet. Made pFC9-T7-Bisulfite and pFC9-T7 (refered to as sample 1+2) as one sample with total volume of 40 ul since IVT will not differ for these samples.
- Incubate all samples for 20 mins at 37C in thermocycler. After incubation added EDTA to final concentration of 24 mM in both samples (4x concentration of MgCl2 in reaction buffer).
- Incubate samples on ice for 10 minutes.
- Add 200 ul npH20 to each sample to increase volume. Then added 1 volume phenol to each sample and added to phenol spin column. Centrifuge each sample at 13,500 rpm for 10 minutes. Removed top layer to new tube.
- Add 2.2 volumes EtOH, 0.1 volumes sodium acetate and 2 ul glycogen to each sample. Froze all samples in -80C for 10 mins.
- Centrifuged at 14,000 rpm for 30 mins.
- Remove supernatent and wash twice with 70% EtOH. Centrifuge at 15,000 rpm for 5 minutes between washes.
- Dry pellet for 10 minutes.
- Add 40 ul npH20 to sample 1+2 and 20 to untranscribed control.
- Split sample 1+2 into two 20 ul samples; pFC9-T7-Bisulfite and pFC9-T7.
- Took 0.5 ul each sample and combined with 9.5 ul npH20 and 1.8 ul purple loading dye. Loaded all samples onto 0.8% 1x TBE gel. Run for 2 hours at 120V.
IVT agarose gel #
Took 0.5 ul from each sample and ran on 0.8% 1x TBE gel. Post-stained gel for 1 hour in 1 ul / 100 ml EtBr. Miscalculated the amount of DNA each sample contained. If no DNA was lost at any step then each sample would have only had 35 ng of DNA (1400 per sample in 20 ul volume = 70 ng/ul).

12/17/21 #
IVT for bisulfite plus EDTA test #
Wanted to re-run the IVT for bisulfite conversion protocol I outlined in 12/15/21 notes and test if the EDTA concentration I was using was sufficient to completely abolish T7 activity. Ran 12/15/21 protocol with 1400 ng pFC9 per sample. Had three samples; pFC9, pFC9 + T7, pFC9 + T7 + 24 mM EDTA. All samples incubated at 37C for 20 mins after which I added 0.96 ul EDTA to sample 2 (pFC9 + T7) and incubated all samples on ice for 5 mins.
After completing Phenol and EtOH precipitation resuspended samples in 20 ul TE and ran half that volume on a 0.8% agarose gel made with 1x TBE. Ran gel in 1x TBE at 60V for 2 hours and then post-stained with EtBr at a concentration of 1.5 ul/ 100 ml EtBr for 1.5 hours (during lab meeting) and then imaged. Gel is shown below.
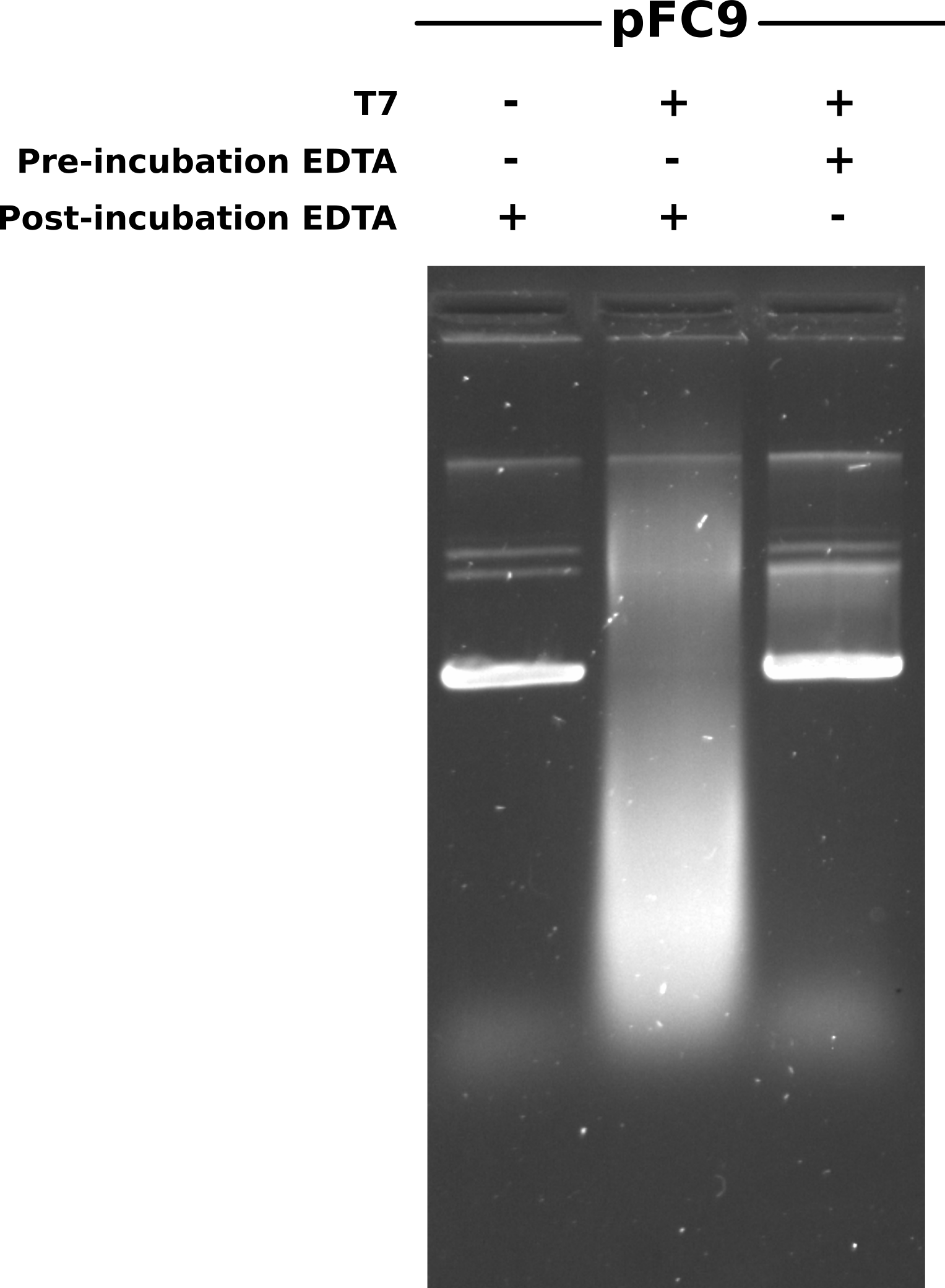
First impression is what happened with the transcribed then EDTA sample (lane 2) that caused such a smear of DNA? Some kind of degradation? Second is that for the most part EDTA appears to have prevented transcription (lane 1 vs lane 3). However it still looks like some transcription has occurred so I plan to increase EDTA concentration from 4x to 5x MgCl concentration.
Looking back, I did not do an RnaseA digestion for any of the samples. The smear is likely due to RNA. Just from looking at the mechanism of RNaseA, no Mg is required. So should be be able to incubate with polymerase, kill with EDTA, digest with RnaseA and then phenol / EtOH precipitate the DNA.
12/21/21 #
IVT run with T7 init midi preps #
Ran through IVT for bisulfite protocol with midi preps of T7 init series. I set up reactions using 2 ul of each insert. The table below describes the reagents for each reaction. The samples described in the table below were made and then split into 2 20ul aliquotes. I then added 2ul T7 polymerase to one and incubated all samples at 37C for 20 mins in the hot room.
| Template DNA name | Concentration (ng/ul) | DNA (ul) | 10X Transcription buffer | DTT | rNTP | npH20 |
|---|---|---|---|---|---|---|
| pFC9 | 300 | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR10 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR11 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR12 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR13 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR17 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR19 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR20 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR21 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR23 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR25 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR26 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR27 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR28 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR29 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
| pFC9VR30 | UNKNOWN | 2 | 4 | 4 | 0.8 | 29.2 |
After incubation added EDTA to all samples to a final concentration of 30 mM (5x transcription buffer concentration of MgCl2). I then added 1 ul 1:1000 dilution of 1mg/ml RnaseA to all samples and incubated for 0.5 hours at 37C in the hot room. After RnaseA incubation I phenol / EtOH precipitated the transcribed samples using phase lock columns for phenol precipitation. Un-transcribed samples were placed on ice.
After completing phenol / EtOH precipitations (2 washes 70% EtOH) I dried all pellets for ~ 1 hour and then re-suspended in 20 ul 10 mM Tris HCL,vortexed and then quick spin. The total volume was more like 40 ul for all samples (possible error with the serial pipetter?) and so I then aliquoted 20 ul of each transcribed sample and 10 ul of the un-transcribed into PCR tubes. Added an additional 10 ul npH20 to the untranscribed samples so volumes of all samples were the same and then added 3.6 ul purple loading dye to all samples.
Agarose gel #
Gel preparation #
Made 1x TBE gel with 0.8% agarose. No EtBr in the gel or the running buffer. Running buffer also 1x TBE.
Sample layout #
Lanes were from right to left in order of ascending insert number. Two lanes per insert with the rightmost being the untranscribed control and the leftmost being the T7 treated. Total of 32 loaded wells.
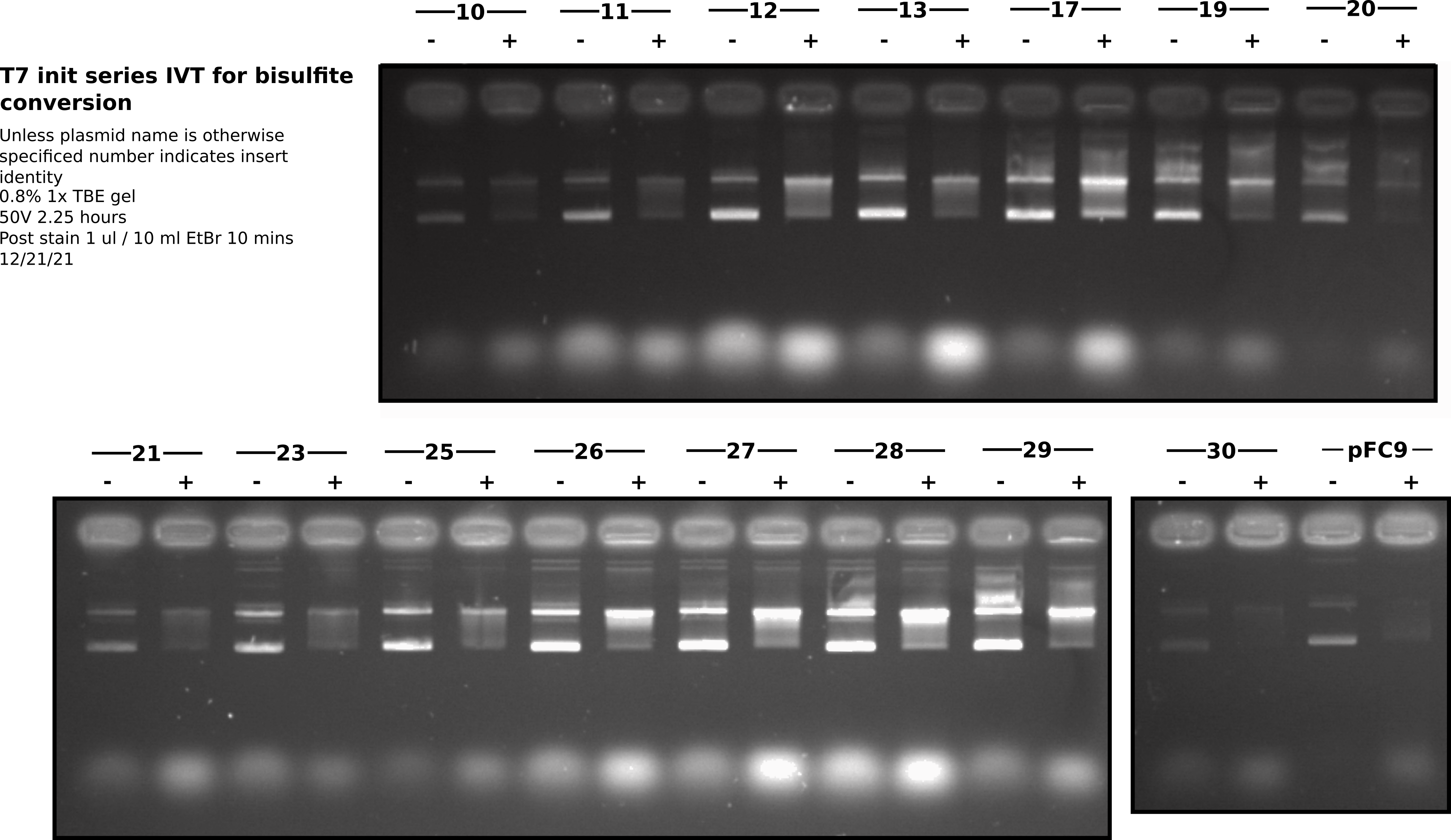
All samples were run on the same gel. Each row of wells is shown as a different section. Overall I think this gel looks very nice. However, there does seem to be a bit less variability between inserts than I had seen in preliminary IVT’s with mini preps. Maybe this prep was more consistent or the midi prep DNA is more consistent in some way (salt, residual proteins etc.). It could also be that EDTA did not completely kill transcription and so all samples continued transcription during RnaseA digestion (1/2 hour) leading to even poor initiators forming R-loops. In the future it may not be necessary to do the phenol extraction on un-transcribed samples but should at least do the EtOH precipitation in order to make sure final volumes are consistent.