sgRNA synthesis and validation #
sgRNA synthesis and validation spreadsheet #
All validation experiments and synthesis details can be accessed at this link.
1/20/22 #
T7_init_VR-13_0-01-18-2022 sgRNA synthesis: start #
Followed protocol for sgRNA synthesis using EnGen kit with T7_init_VR-13_0-01-18-2022 oligo. Only deviation was I started
Phenol-chloroform purification before DnaseI digestion. So I completed
phenol-chlorform purifcation, resuspended in 45 ul npH20, added 5 ul 10x DnaseI
buffer and 1 ul DnaseI. Completed digestion and then preformed another
phenol-chloroform purification stopping at 100% EtOH precipitation freezing
samples overnight.
1/21/22 #
T7_init_VR-13_0-01-18-2022 sgRNA synthesis: finish #
Completed EtOH precipitation and resuspended sample in 50 ul 0.1 mM EDTA. Measured concentration using nanodrop at 548 ng/ul.
T7_init_VR-13_0-01-18-2022 sgRNA: validation #
Prepared samples according to table below in order to test sgRNA and Cas9 nickase activity with it.
| Nuclease free H20 | 10X NEBuffer r3.1 Reaction Buffer | sgRNA species | Diluted sgRNA volume (ul) | EnGen Spy Cas9 Nickase (M0650) (ul) | Template DNA (ul) | Template DNA mass (ng) | Sample Number |
|---|---|---|---|---|---|---|---|
| 37.66010645 | 6 | pFC9VR13Cas9-0 | 10.03719088 | 2 | 4.302702673 | 391.5459432 | 3 |
| 47.69729733 | 6 | pFC9VR13Cas9-0 | 0 | 2 | 4.302702673 | 391.5459432 | 2 |
| 49.69729733 | 6 | pFC9VR13Cas9-0 | 0 | 0 | 4.302702673 | 391.5459432 | 1 |
Complete reaction details available at this link.
For all samples followed NEB in vitro Cas9 protocol.
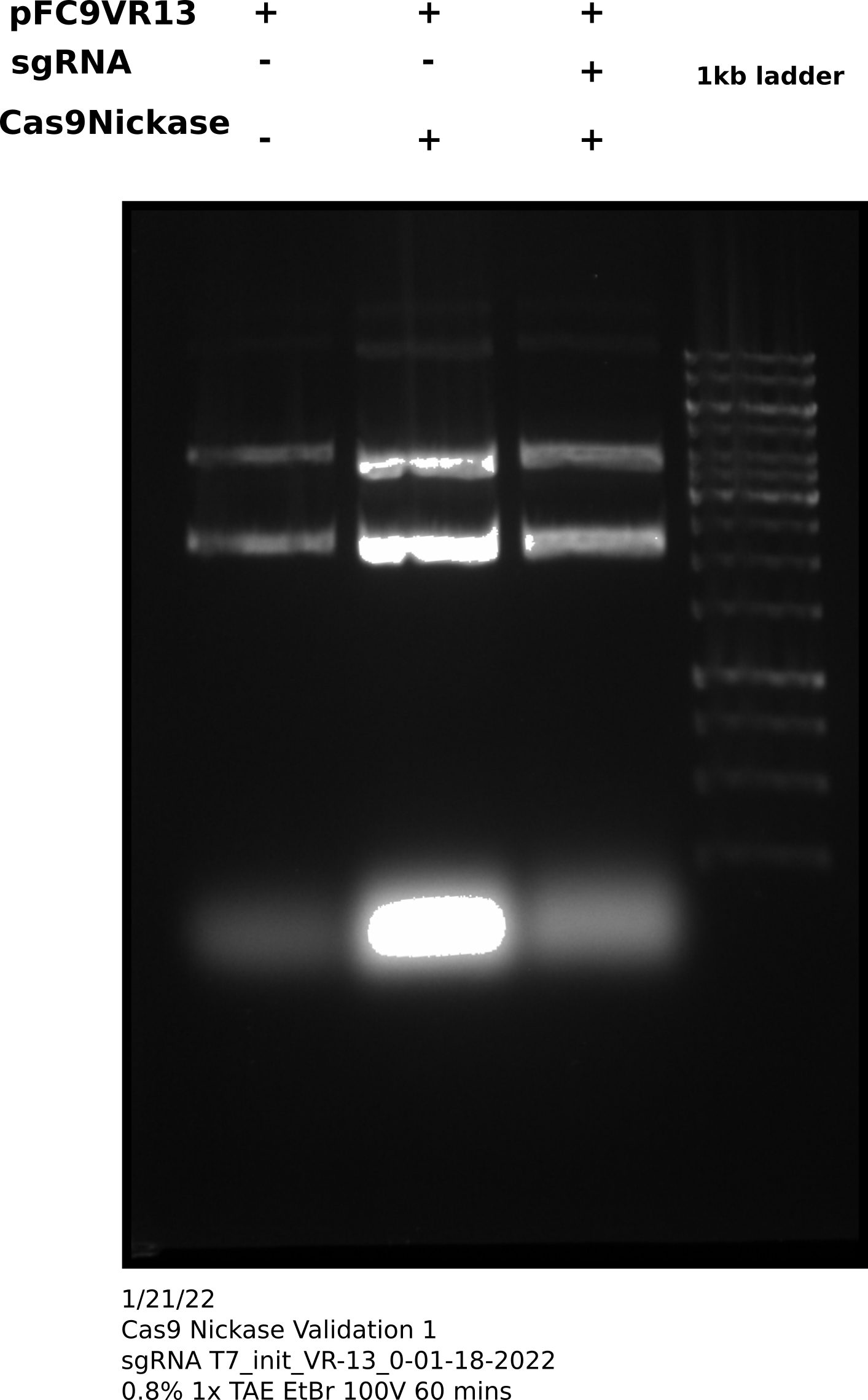
After completing protocol ran 33 ul of each sample (~200 ng) each sample on agarose gel shown in the image below. Saved the remaining samples (27 ul) at 4C.
Cas9 digestion agarose gel sgRNA 0 #
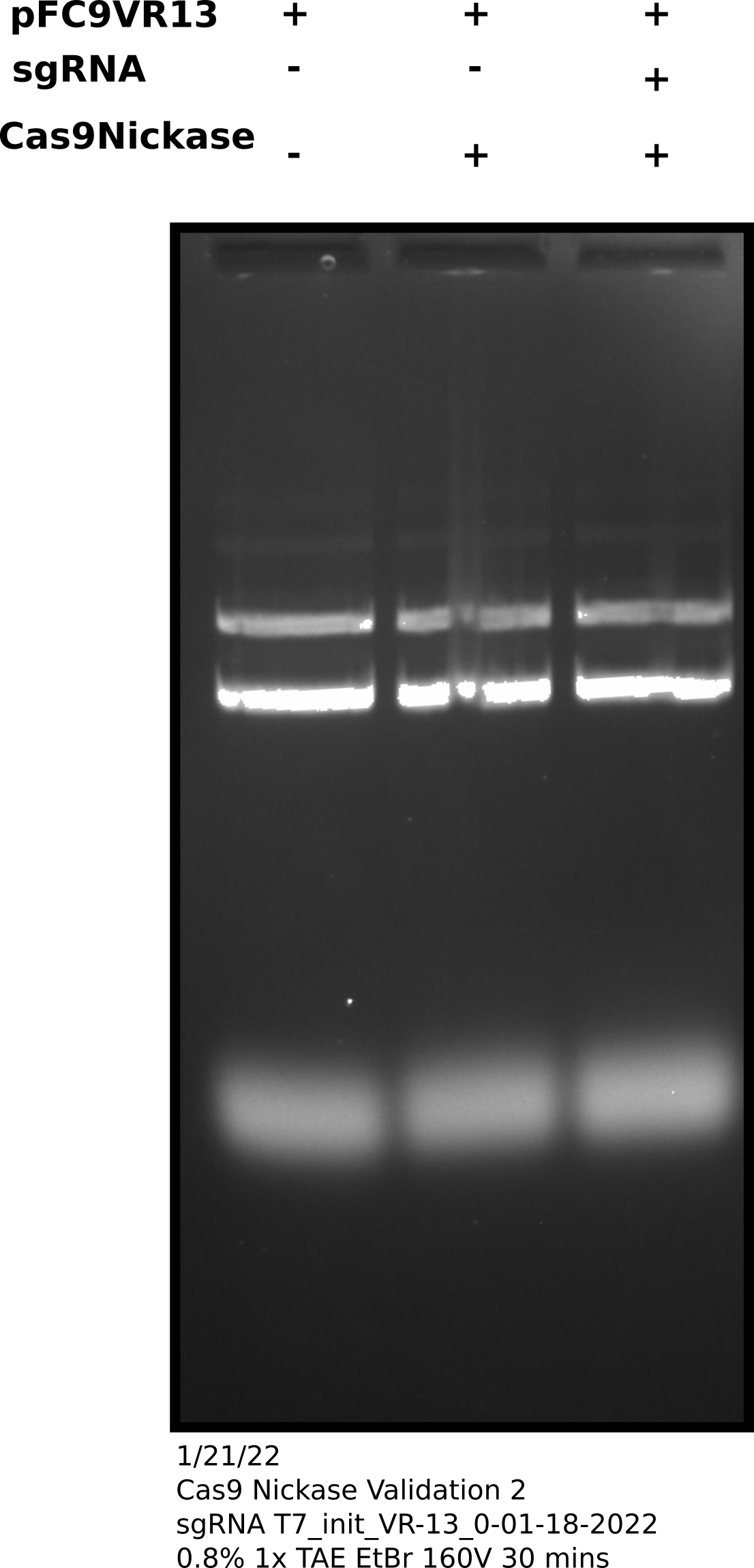
Cas9 digestion agarose gel sgRNA 0: corrected Cas9 concentration #
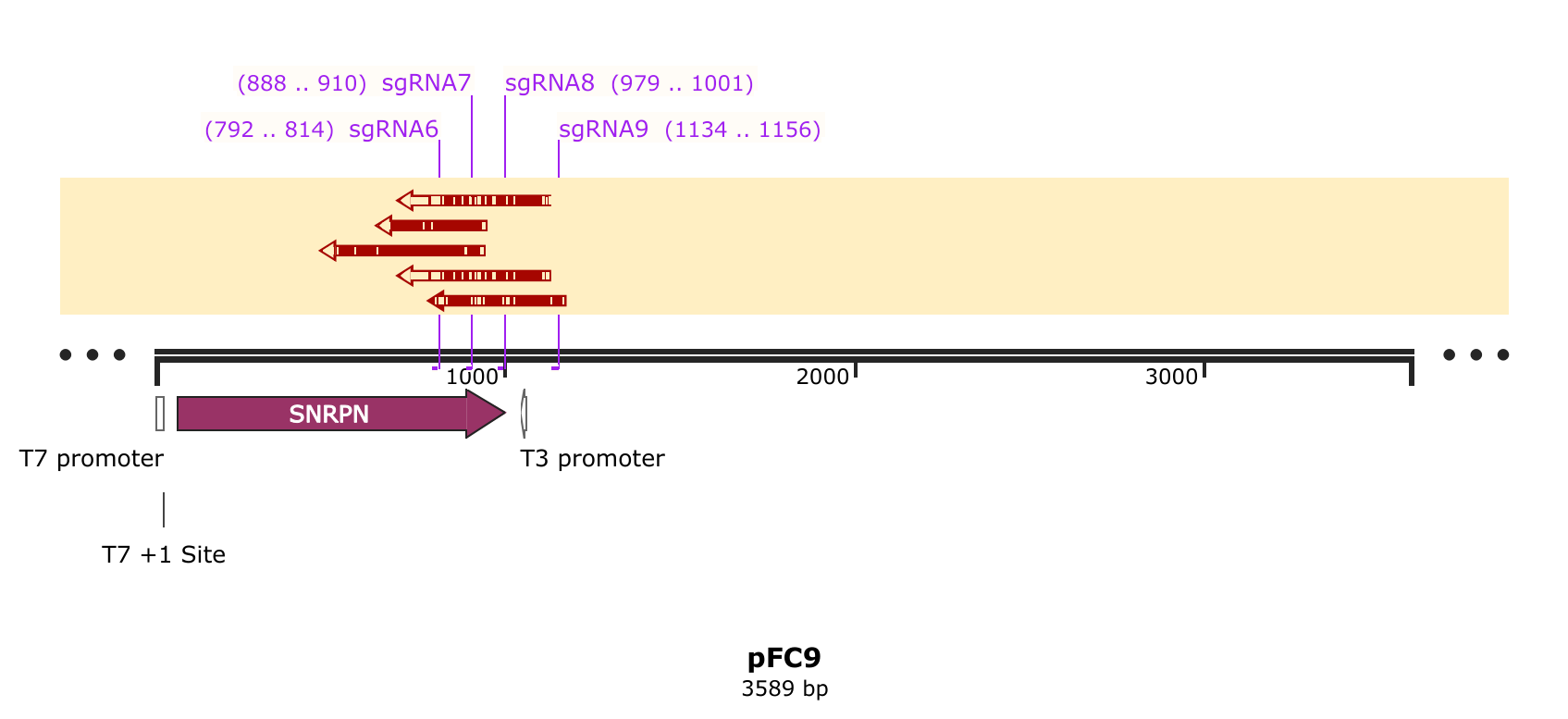
Oligo design triple check #
Since I suspect guide RNA is the issue I double checked my sgRNA oligo generating workflow using one of the VR13 oligos I ordered. First I checked the target sequence to make sure it is present in pFC9VR13.

Snapgene alignment using the target sequence as a primer confirms target sequence presence. Next, I wanted to make sure the oligo the workflow generated matches what NEB would say to order for their EnGen synthesis kit. So I copied the target sequence into NEB sgRNA oligo design tool; this produced the output shown in the image below.
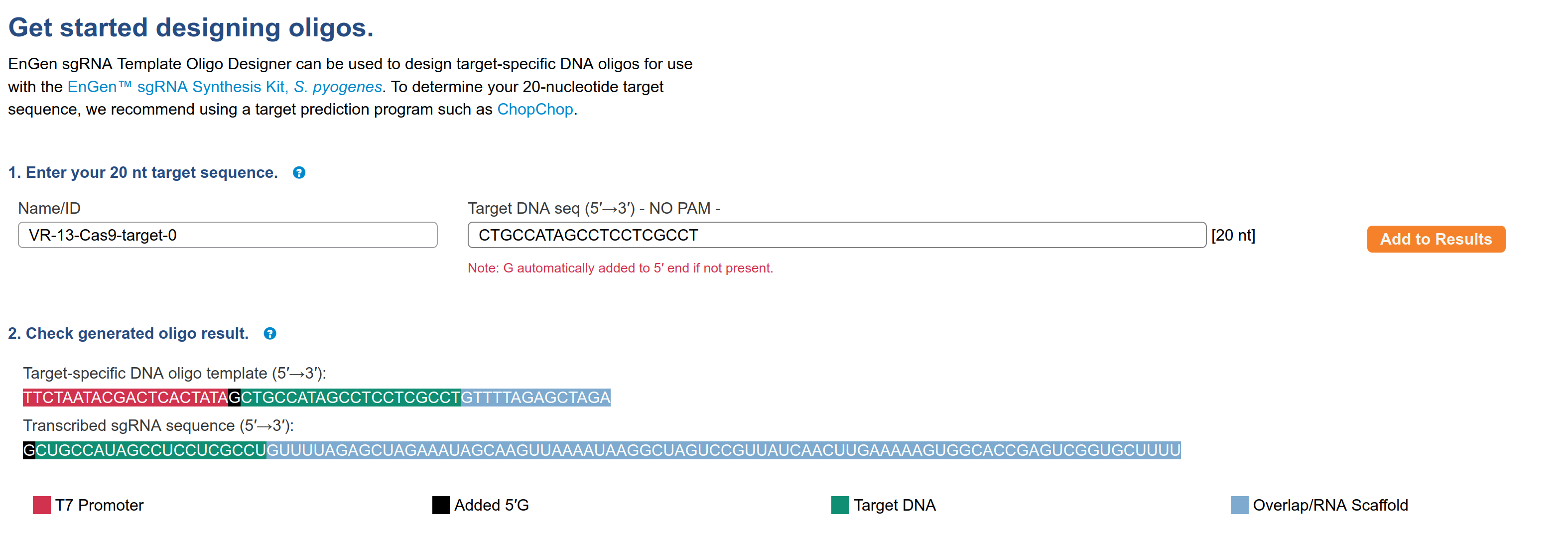
The sequence of the oligo is
TTCTAATACGACTCACTATAGCTGCCATAGCCTCCTCGCCTGTTTTAGAGCTAGAThis exactly matches what the workflow produced for this target sequence
in the output/IDT-order/T7_initiation_series/T7_init_VR-13.NEB.selected.targets.scored.IDT.tsv
file and is shown below along with other fields generated.
T7_init_VR-13_0-01-18-2022 TTCTAATACGACTCACTATAGCTGCCATAGCCTCCTCGCCTGTTTTAGAGCTAGA 25nm STDAnd finally I wanted to check this oligo was actually what I told IDT to make for me. So I went back to my order history and pulled up the sequence order.
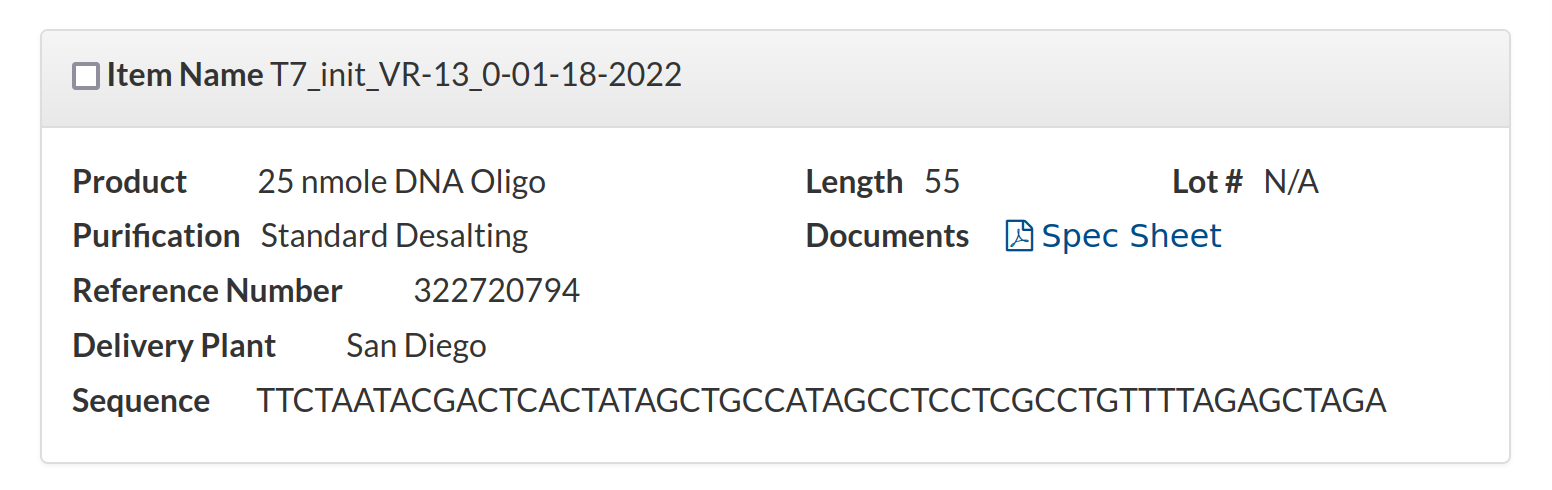
The sequence shown here is
TTCTAATACGACTCACTATAGCTGCCATAGCCTCCTCGCCTGTTTTAGAGCTAGAso everything matches and is targeted correctly. At this point I am confident sgRNA design is not the problem.
1/22/22 #
Cas9 sgRNA synthesis #
Used NEB EnGen kit and protocol with remaining ordered but un-synthesized oligos (table below).
| Oligo name |
|---|
| T7_init_VR-13_1-01-18-2022 |
| T7_init_VR-13_2-01-18-2022 |
| T7_init_VR-13_3-01-18-2022 |
| T7_init_VR-13_4-01-18-2022 |
After completing synthesis used nanodrop to measure concentrations of all samples, shown in table below.
| sgRNA | ng/ul | 260/280 | 260/230 |
|---|---|---|---|
| T7_init_VR-13_1-01-18-2022 | 856.6 | 1.616 | 2.15 |
| T7_init_VR-13_2-01-18-2022 | 1621 | 1.813 | 2.396 |
| T7_init_VR-13_3-01-18-2022 | 911.6 | 1.832 | 2.368 |
| T7_init_VR-13_4-01-18-2022 | 864 | 1.732 | 2.272 |
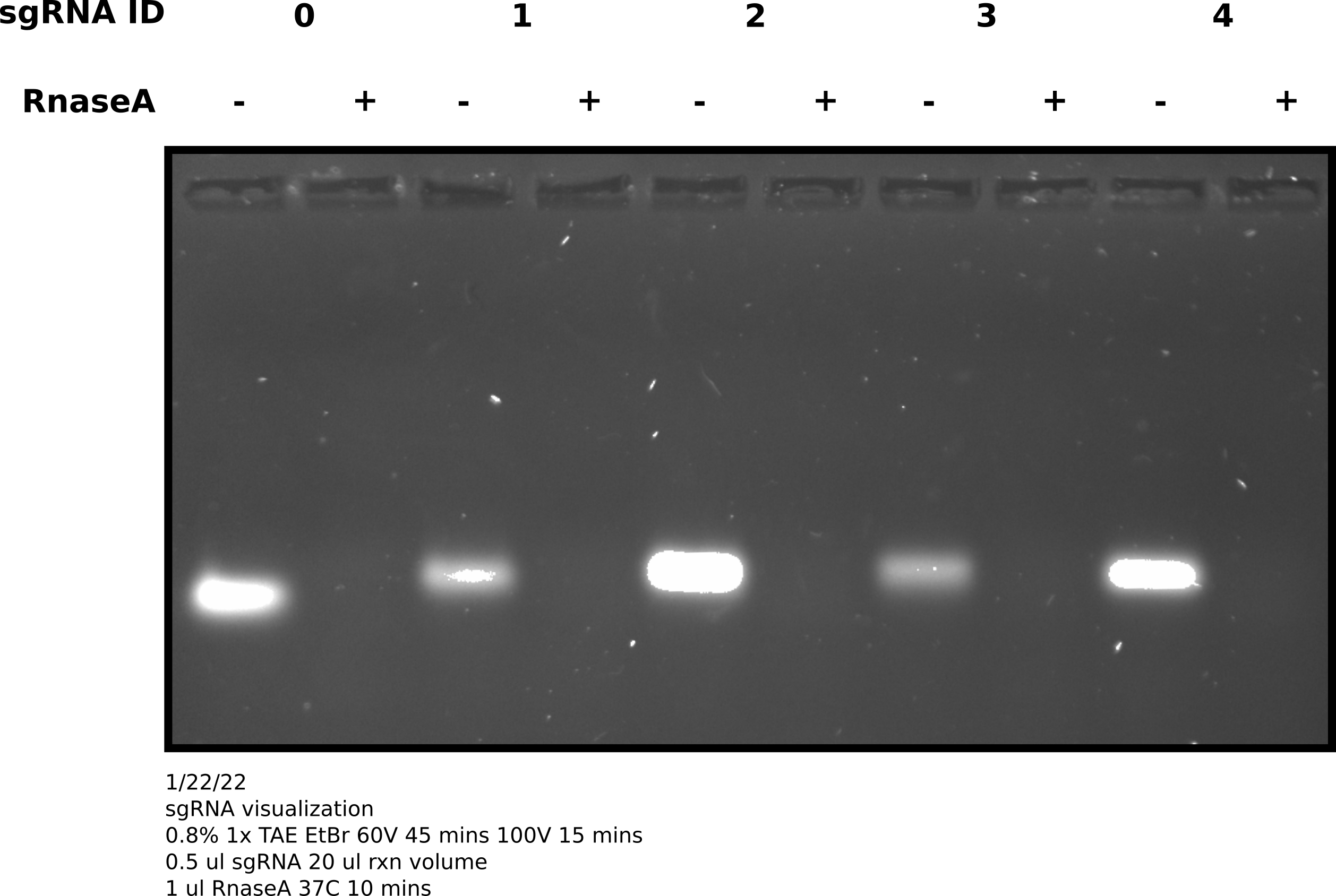
0 may be running slightly
faster but it is hard to tell. Would have been better to run this on a
higher percentage gel but for now this confirms sgRNA synthesis is producing
sgRNAs of ~ the sample length.1/23/33 #
Cas9 + sgRNA validation test 2 #
Since I know sgRNAs are def RNA and have a mostly consistent length I am testing if sgRNA 0 just happened to be a lemon. I set up reactions according to the table below, which is also available at this link along with sgRNA concentration information. I used pFC9 as the template DNA as it shares all target sites with pfC9VR13 which was used for the actual template for these guides. I used pFC9 instead of pFCVR13 because the pFC9 sample has the same target sites but there is significantly less contaminating E. coli RNA present; which could be pointed to as a confounding variable for Cas9 activity.
| Nuclease free H20 | 10X NEBuffer r3.1 Reaction Buffer | sgRNA species | Diluted sgRNA volume (ul) | EnGen Spy Cas9 Nickase (1uM) | Template DNA (ul) | Template DNA mass (ng) | Sample Number |
|---|---|---|---|---|---|---|---|
| 52.69484686 | 6 | NA | 0 | 0 | 1.305153144 | 391.5459432 | 1 |
| 40.02410197 | 6 | pFC9VR13Cas9-0 | 10.67074489 | 2 | 1.305153144 | 391.5459432 | 2 |
| 43.93933831 | 6 | pFC9VR13Cas9-1 | 6.755508549 | 2 | 1.305153144 | 391.5459432 | 3 |
| 47.08746364 | 6 | pFC9VR13Cas9-2 | 3.60738322 | 2 | 1.305153144 | 391.5459432 | 4 |
| 44.28022619 | 6 | pFC9VR13Cas9-3 | 6.414620667 | 2 | 1.305153144 | 391.5459432 | 5 |
| 43.92682811 | 6 | pFC9VR13Cas9-4 | 6.76801875 | 2 | 1.305153144 | 391.5459432 | 6 |
I manually confirmed that all target sequences (and PAM sites) used when designing these oligos are present in pFC9 using snapgene. The plasmid map below shows the target sequences + PAM sites mapped onto pFC9.
I followed NEB EnGen in vitro digestion protocol with all samples. After incubation with Cas9 saved 40ul of sample for later use and treated the remaining sample with 0.66 ul ProteaseK for 10 mins at room temp. After treatment added 3.33 ul purple loading dye and ran samples on 0.8% agarose gel.
sgRNA comparison agarose gel #
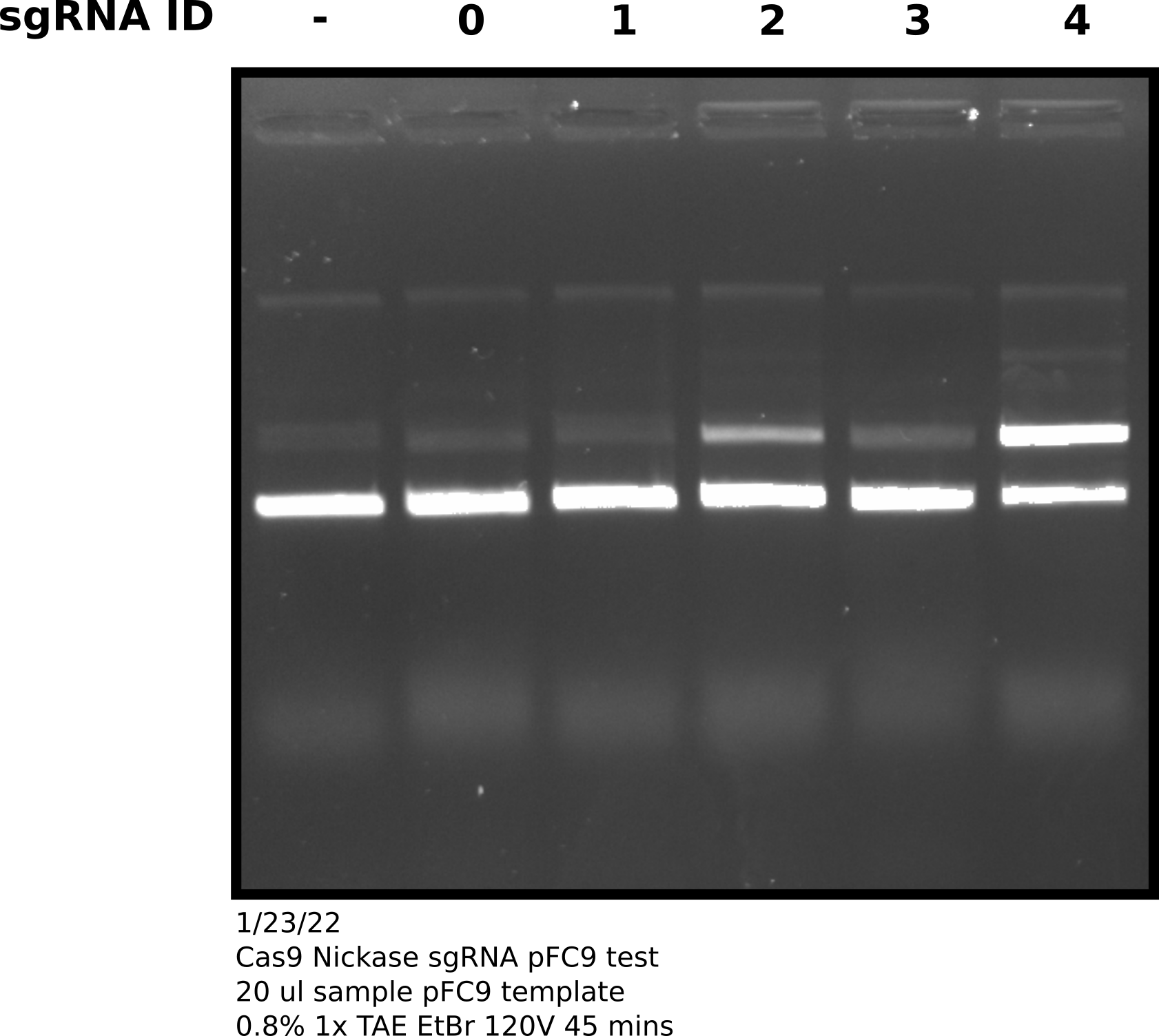
However, after talking to Fred about these results he pointed out that the sgRNA comparison gel shows that the Cas9 enzyme we have is capable of nicking to some degree. It may be the case that more is required to get higher efficiency. This goes somewhat against sgRNA 0 results which showed no activity with the nickase even when I accidentally used 20X the recommended amount of Cas9. Fred suggested attempting to get Cas9 working with sgRNA 4 at high efficiency in order to then better diagnosis other issues (sgRNA effecting nicking efficiency).
1/25/22 #
Cas9 nickase and sgRNA4 titration #
In order to get a better idea what may be the limiting reagent I set up
a 2D titration of sgRNA4 (oligo ID:oEH19 and the best preforming sgRNA based on yesterday’s results) and Cas9 nickase.
The detailed reaction setup tables can be found
at this link. In summary,
I setup 4 dilutions of Cas9 and sgRNA of
increasing concentration. I then added a fixed
amount of each of these dilutions in a grid
like manor to a PCR grid in order to create
16 unique conditons. I followed NEB Cas9 in vitro digestion protocol as usual for all protocol related to
reaction setup order and timings. The final concentrations of both sgRNA and Cas9 in all samples ranged from between 1X NEB recommended concentration (30 nM) to 10X that.
After completing the in vitro digestion protocol I added 3 ul purple loading dye to all samples and ran them on an agarose gel. Shown in the image below.
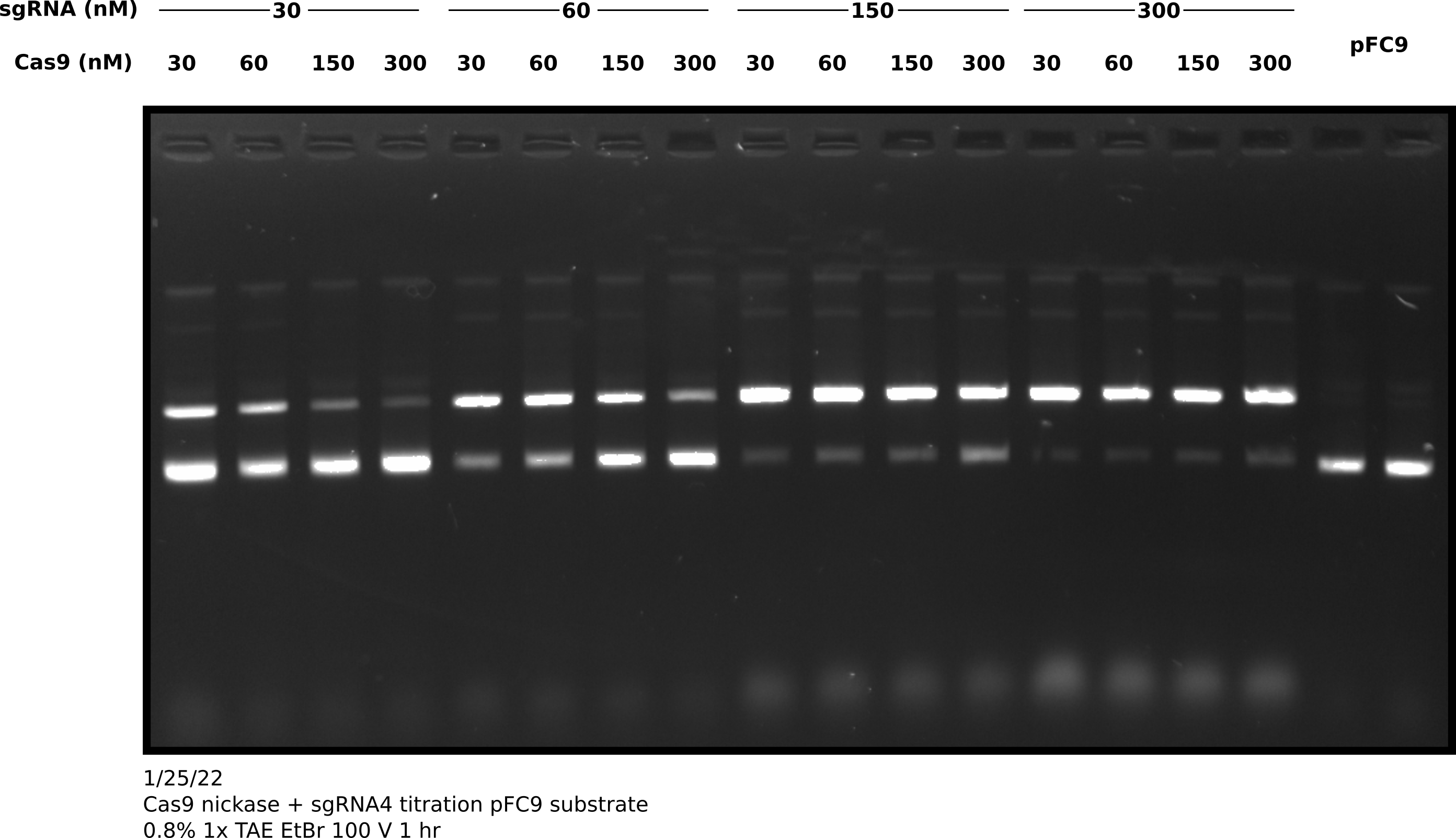
I then quantified this image by comparing the percentage of nicked band between lanes. Quantification code as jupyter notebook at this link.
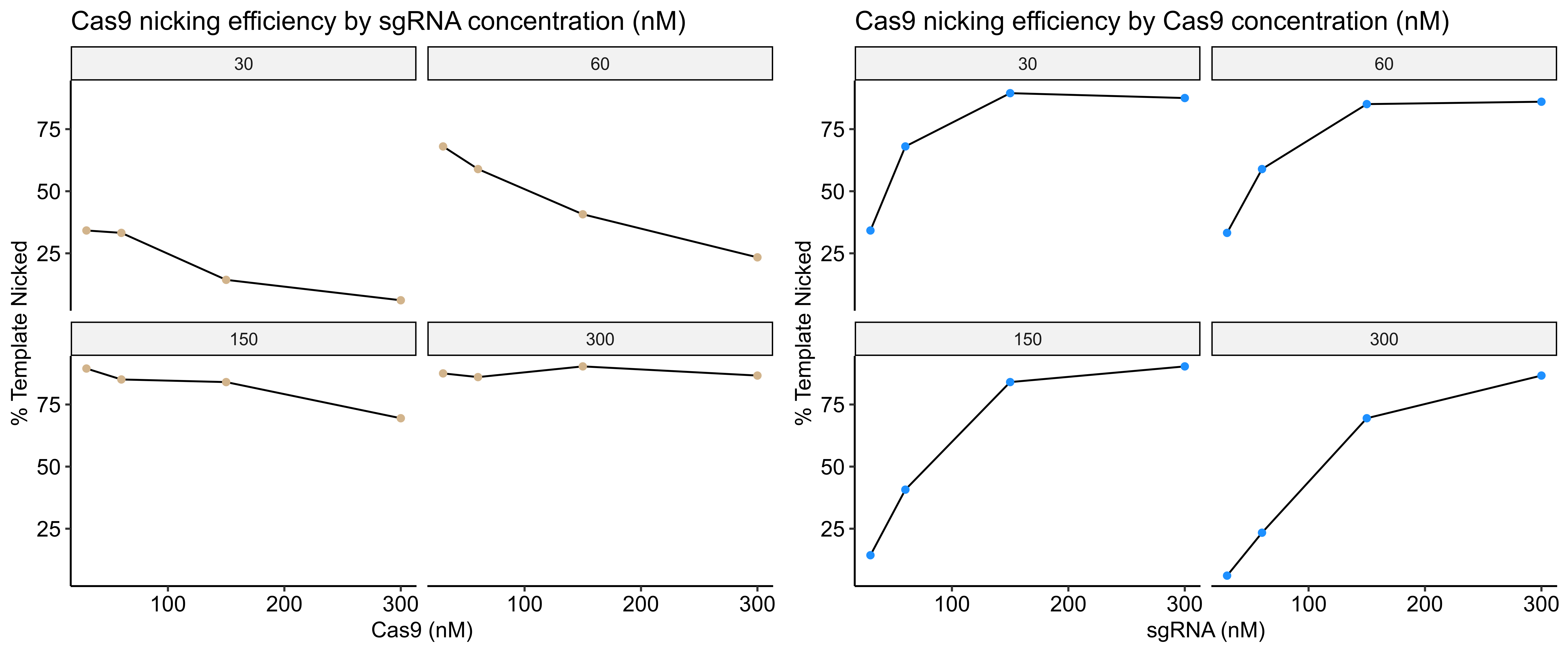
Based visually on the gel and the quantification increasing sgRNA concentration has the most impact on Cas9 nickase efficiency; at least with this particular sgRNA. In fact, high concentrations of Cas9 with relatively low concentrations of sgRNA seem to reduce the efficiency of the reaction.
1/26/22 #
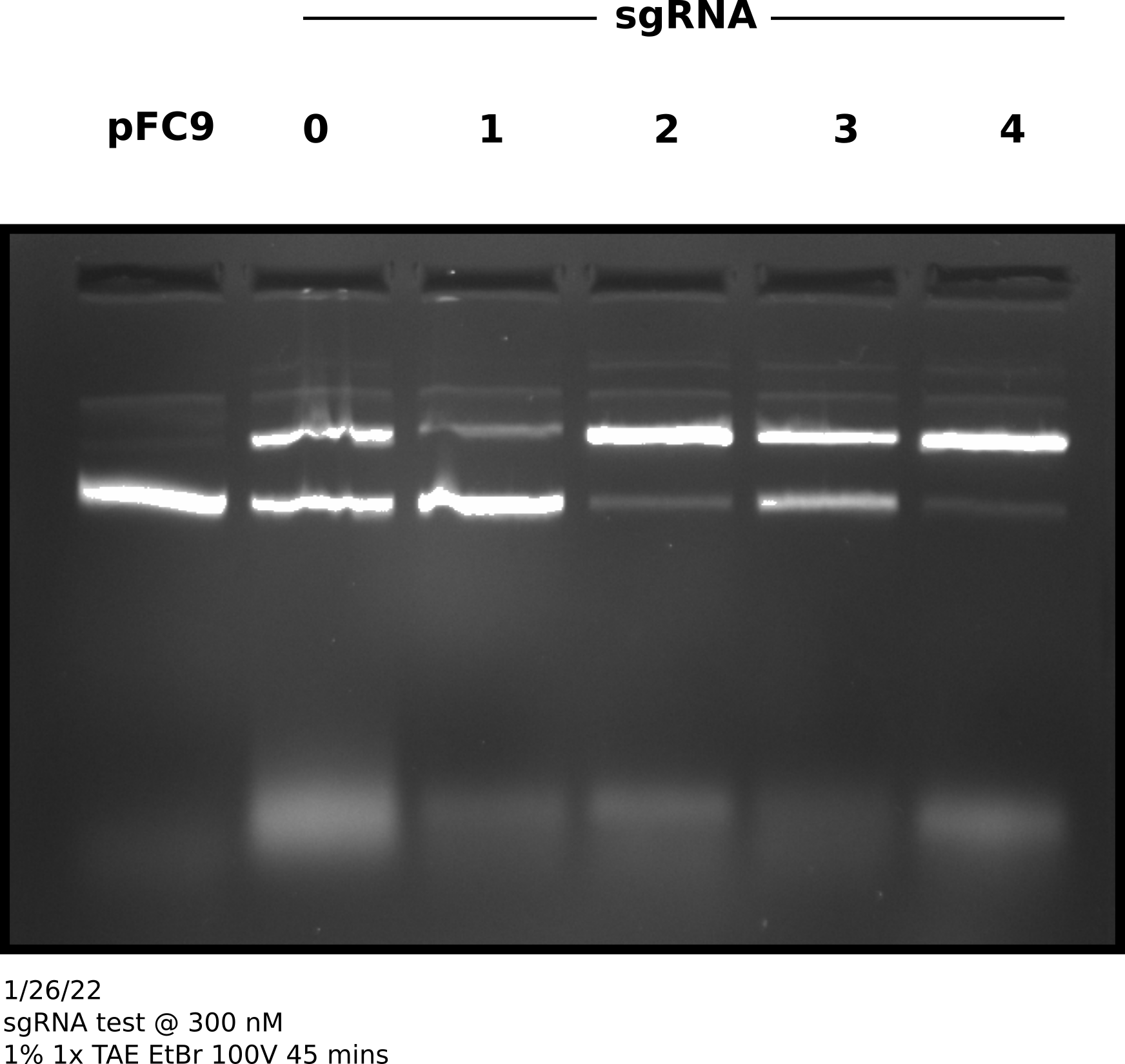
sgRNA test at 300 nM #
Since 300 nM of sqRNA seemed to give much higher nicking efficiencies independent of Cas9 concentration I tested Cas9 nickase efficiency at 30 nM with 300 nM sgRNA and 3 nM template DNA (pFC9) for all currently synthesized sgRNAs. Complete sample prep is described at this link. After following NEB Cas9 in vitro digestion protocol with all samples.
After completing protocol ran entire sample volume on agarose gel at 100V for 45 minutes (shown in image below).
sgRNA 300 nM test gel #
I then quantified this difference as the percent % for each sgRNA at 300 nM - the % nicked at 30 nM sgRNA (link to Jupyter notebook) which is shown in the plot below.
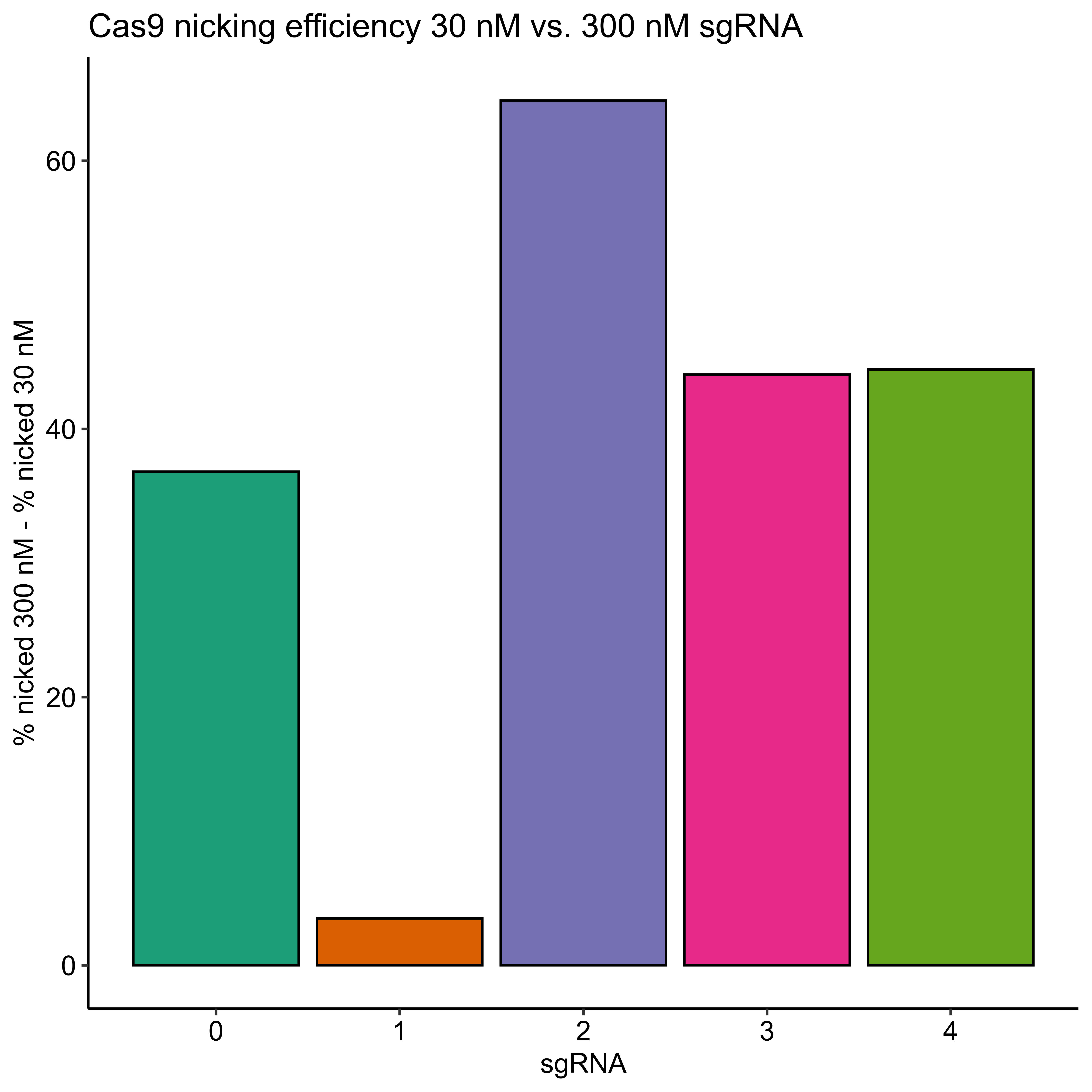
Efficiencies for all sgRNAs except sgRNA 1 did increase but not all to the 90% level. This is indicating to me that the sequence or something else specific to each sgRNA is effecting the efficiency in some way that is significant in at least some cases (sgRNA 1) to negate the effects of increased concentration.
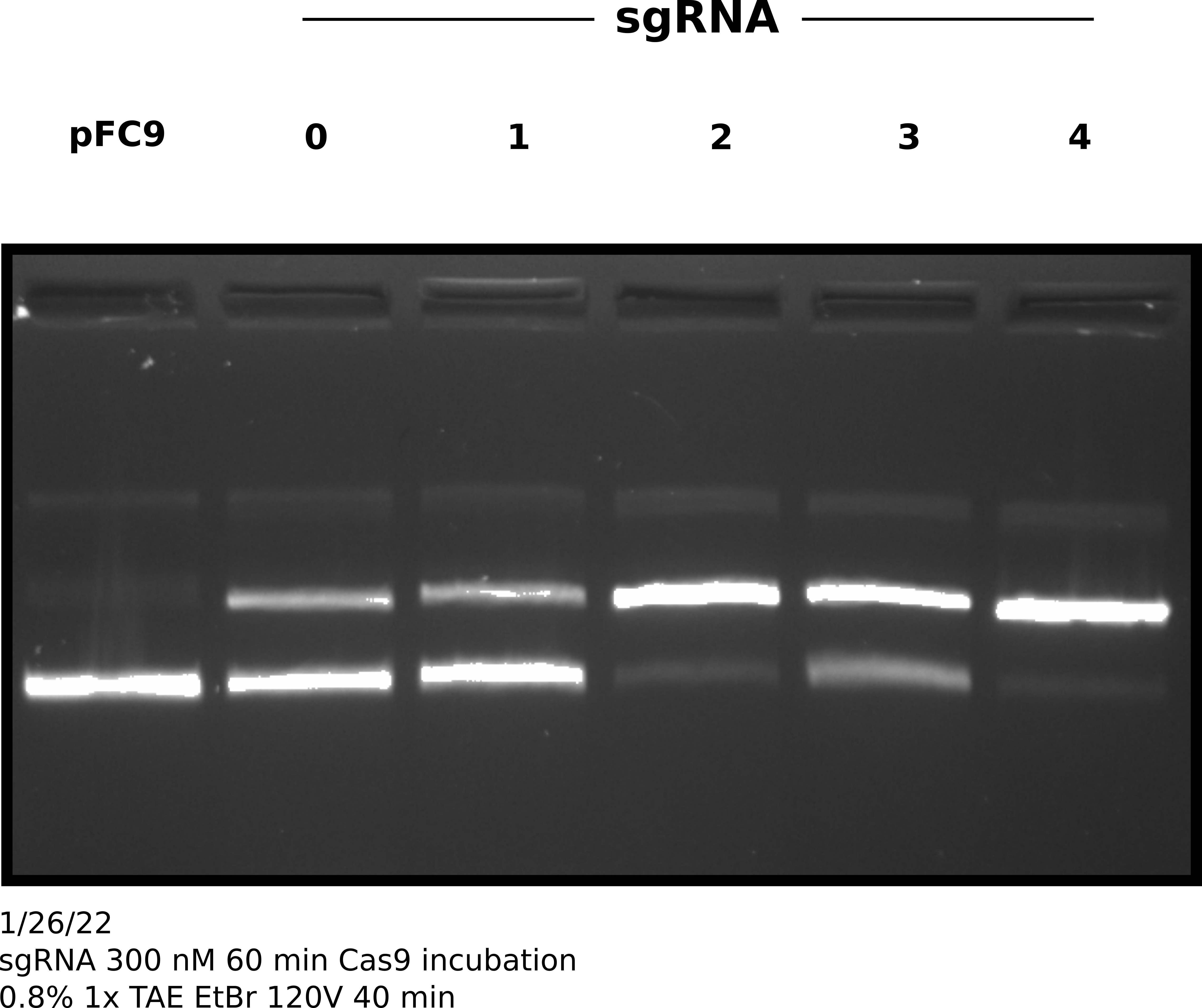
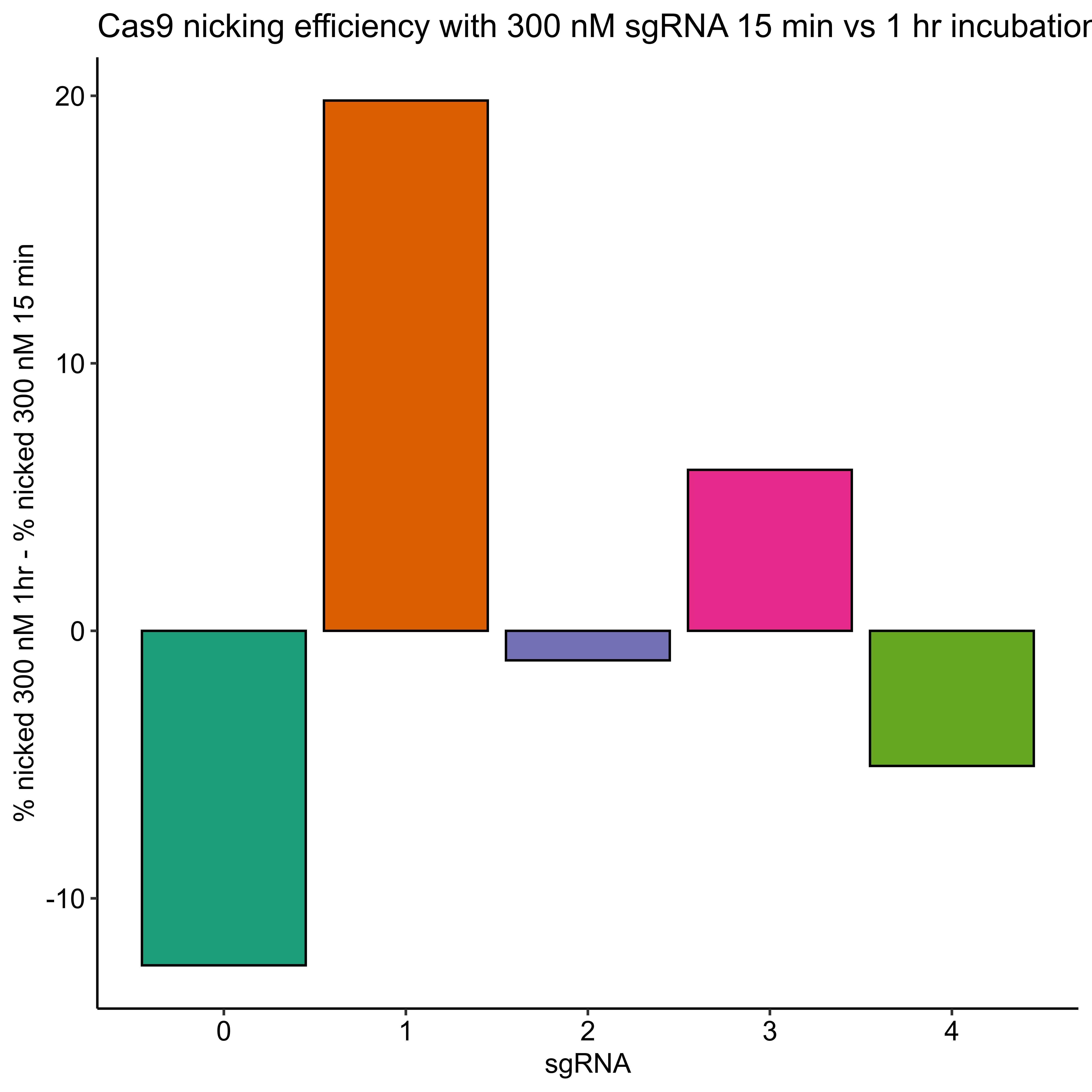
In order to test if longer incubation with Cas9 night up the efficiencies of sgRNAs that are underpreforming I repeated this same experimental setup but increased the incubation time with Cas9 at 37C to 1 hour. After completing this protocol I ran samples out on agarose gel and quantified the results in the same manner.
sgRNA 300 nM test with 1hr 37C Cas9 incubation gel #
Percent template nicked by sgRNA quantification #
2/2/22 #
sgRNA 4 validation via Sanger sequencing: Sample prep #
Since sgRNA 4 has shown high efficiency after optimization I went ahead and set up an in vitro digestion of pFC9 using this sgRNA to validate via Sanger sequencing at the Davis Sanger center.
Experimental setup and protocol #
I prepared samples and dilutons according to the table below, which is also available at this link.
Samples were prepared and treated according to NEB Cas9 in vitro digestion protocol. After completion of the protocol samples were phenol-chloroform extracted using phase lock columns and EtOH precipitated with 2 ul of molecular grade glycogen. All samples were then re-susended in 6 ul 10 mM Tris HCl and boiled at 100C for 5 mins in the thermocycler. After boiling samples were spun briefly in desktop centrifuge and then immediately placed into the freezer.
I was unable to sequence the samples as their were no remaining spots in the sequencer by the time I completed sample preparation. In the same PCR tube strip as the three samples I prepared 20 ul of 3 uM of oEH19_Cas9_validator (oEH24) which targets a region on pFC9 about 200 bp upstream of the sgRNA4 target site.
Sequence submission and analysis #
I submitted sequences on Friday and was returned the results on Monday. I analyzed them using the casanger workflow and produced the plot shown below.
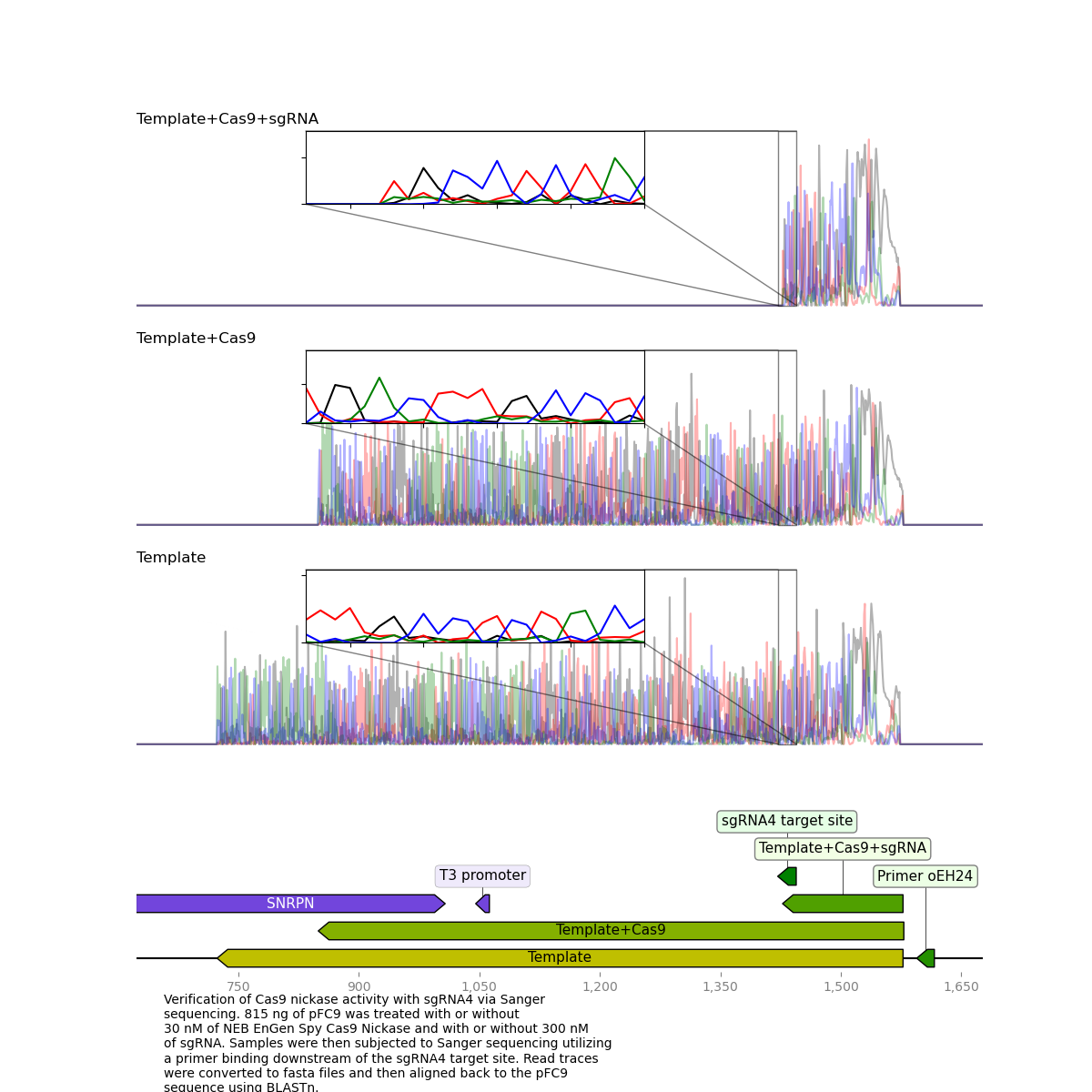
It is very clear that the Cas9 + sgRNA treated template read dies exactly at the target site which was exactly what I was hoping to see.
2/7/22 #
sgRNA 5-9 synthesis #
Prepared sgRNAs 5-9 following sgRNA synthesis protocol. After completing synthesis and purification I used the nanodrop to measure samples concentrations; shown in the table below.
Sample concentrations #
| sgRNA | sgRNA concentration (ng/ul) | Final volume (ul) | 260/280 | 260/230 |
|---|---|---|---|---|
| sgRNA 5 | 842.8 | 100 | 1.883 | 2.378 |
| sgRNA 6 | 899 | 100 | 1.909 | 2.408 |
| sgRNA 7 | 1065 | 100 | 1.911 | 2.401 |
| sgRNA 8 | 995.7 | 100 | 1.925 | 2.447 |
| sgRNA 9 | 937 | 100 | 1.921 | 2.397 |
After measuring concentrations I prepared 1 ul of each sample in 19 ul npH20 and split these aliquots into 2 separate 10 ul samples. I added 1 ul stock RnaseA to one sample (Rnase +) and incubated all samples at 37C for 10 mins after which I added 2 ul purple loading dye. I then ran 10 ul each sample on the gel shown below.
sgRNA 5-9 agarose gel #
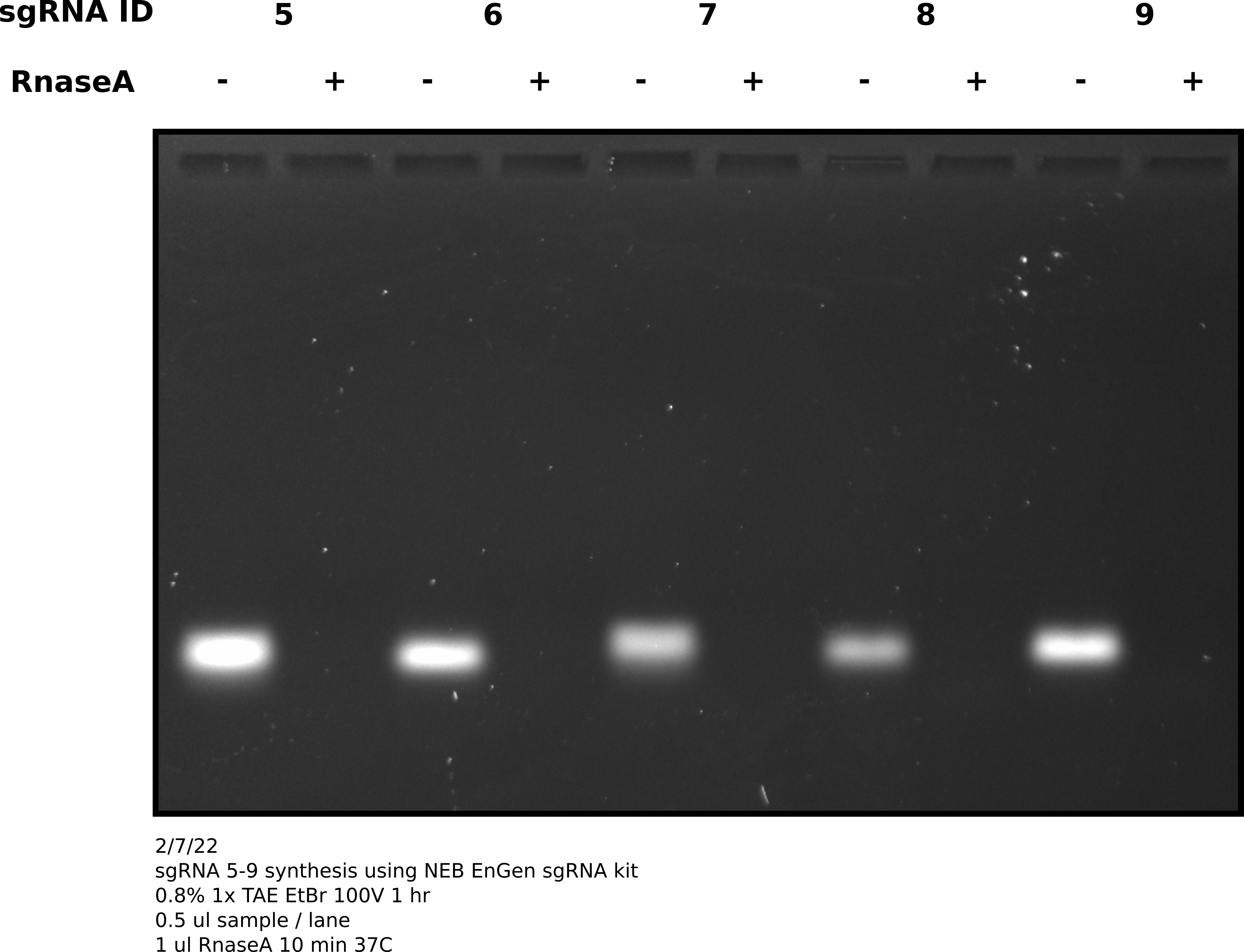
As expected all sgRNAs appear at about the same height and are clearly RnaseA sensitive.
2/8/22 #
sgRNA 5-9 validation #
Since sgRNA yesterday synthesis was successful I moved forward with validating sgRNA activity and specificity. I preformed in vitro digestion of pFC9 template following NEB protocol but scaling reagent volumes linearly and with 300 nM final concentration of each sgRNA. I digested ~815 ng of pFC9 per reaction. This was a sufficient amount to visualize on a gel and use remaining sample for sequencing. Complete reaction setup details are available at this link. After completing in vitro digestion I preformed standard phenol-chloroform followed by EtOH precipitation protocol to remove protein and concentrate samples, re-suspending in a final volume of 8.15 ul of 10 mM TrisHCl.
sgRNA activity assessment via agarose gel #
After completing in vitro digestion and precipitation I ran 2 ul of each sample on a 1% agarose gel in order to visually access Cas9 activity with each sgRNA.
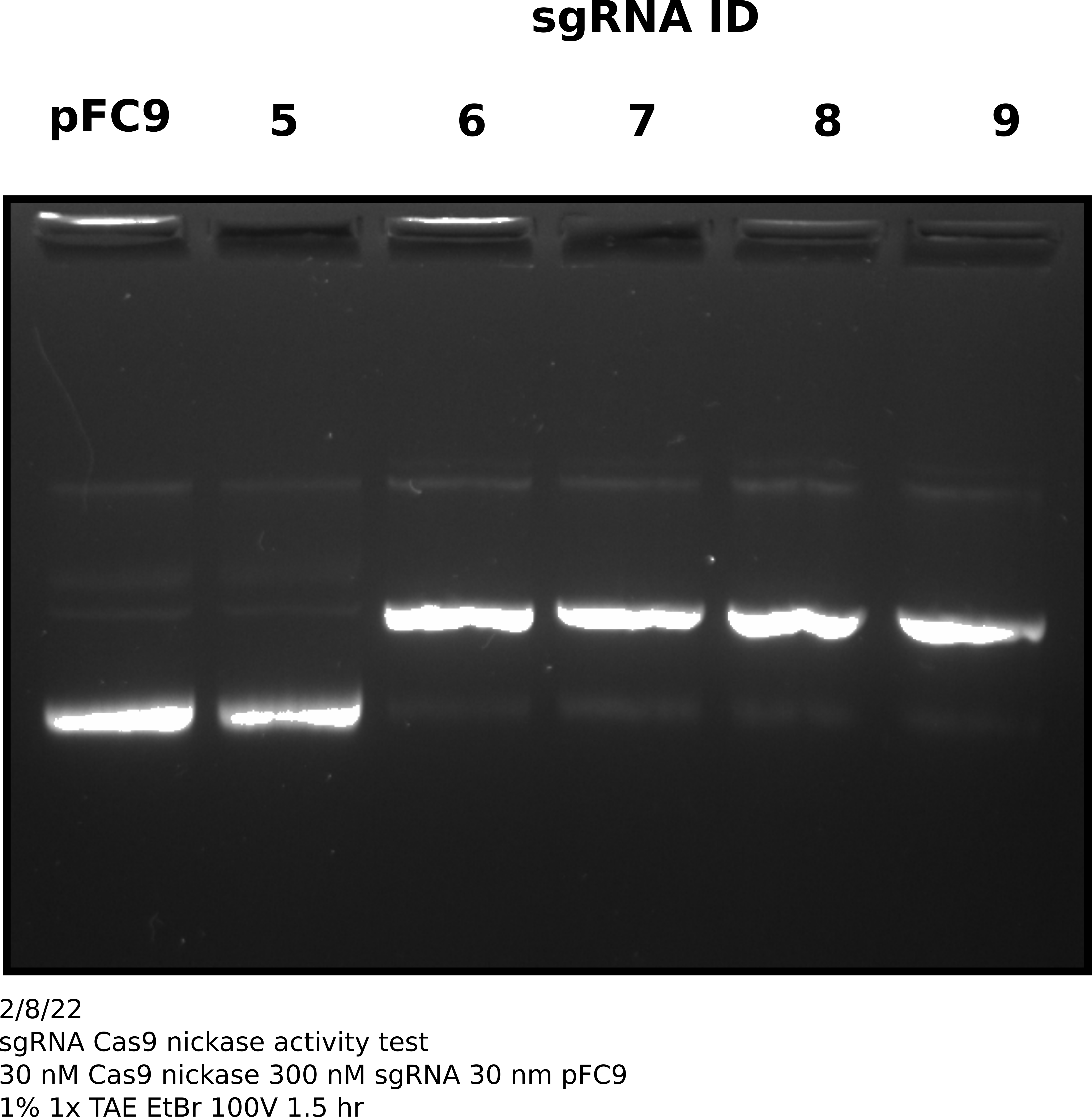
Based on this gel all sgRNAs show very strong activity (>= 90%) except sgRNA 5. Will not sequence sgRNA 5.
Preparation of Cas9 treated negative controls and Sanger sequencing prep #
Note on experiment timing #
These experiments were preformed on 8/9/22 (the day after I completed the reaction described above which are the positive samples). The next day I decided I would also like to have two negatives per sgRNA tested. One with template DNA and Cas9 but no sgRNA and one with neither sgRNA or Cas9 but the same sequencing primer. For continuity of reading I am including the experiment notes here.
Prepared Cas9 treated negative control sample as described by this spreadsheet. Followed same protocol as used for positive samples. After completing in vitro digestion and precipitation I re-suspended sample in 26.10 ul 10 mM Tris HCl and then split this volume into 3 separate PCR tubes for sequencing with each primer. I prepared template only samples with 3.5 ul pFC9 and 3.5 ul npH20. Samples submitted for sequencing at the UC Davis Sanger center are described in the tables below.
Sample submission details #
Sample names, primers and treatments
| Sample name | Primer ID | Cas9 | sgRNA ID | Template |
|---|---|---|---|---|
| 6 + P16 | oEH22 | TRUE | 6 | pFC9 |
| 7 + P16 | oEH22 | TRUE | 7 | pFC9 |
| 8 + P17 | oEH23 | TRUE | 8 | pFC9 |
| 9 + P2 | oEH2 | TRUE | 9 | pFC9 |
| 10 + P16 | oEH22 | TRUE | NA | pFC9 |
| 11 + P17 | oEH23 | TRUE | NA | pFC9 |
| 12 + P2 | oEH2 | TRUE | NA | pFC9 |
| 13 + P16 | oEH22 | FALSE | NA | pFC9 |
| 14 + P17 | oEH23 | FALSE | NA | pFC9 |
| 15 + P2 | oEH2 | FALSE | NA | pFC9 |
Sequencing primer translation table
| Sample submission primer name | Oligo name | oligoID |
|---|---|---|
| P16 | oEH16_Cas9_validator | oEH2 |
| P17 | oEH17_Cas9_validator | oEH22 |
| P2 | pFC8tac_tac_promoter_Primer_2 | oEH23 |
2/9/22 #
sgRNA 5-9 validation continued #
Notes are included under 2/9/22, see this note.
2/11/22 #
sgRNA 5-9 validation: analysis of sequencing results #
Received Sanger traces back from Davis sequencing center and they were not as clean as sgRNA 4 results, despite the degree of digestion as assessed by agarose gel being comparable.
First thing I did was visulize the Cas9 + sgRNA reads aligned against pFC9 using SnapGene as a sanity chekc for any plots produced by my workflow. That alignment is shown in the image below with sgRNA target sites added as primers.
While the quality of reads beyond the nick site is generally not great, there is clearly still a proportion of each read that aligns beyond the each target site. This indicates that some significant amount of un-nicked template is present in these reads. Since assessment by agarose gel of these samples showed >90% nicking efficiency my best bet to what happened was contamination with un-nicked template. This must have occurred after the point where aliquots were taken for the agarose gel.
Looking at individual samples in greater details tells the same story as the overview alignment.
Detailed view: sgRNA 8 traces and alignments #
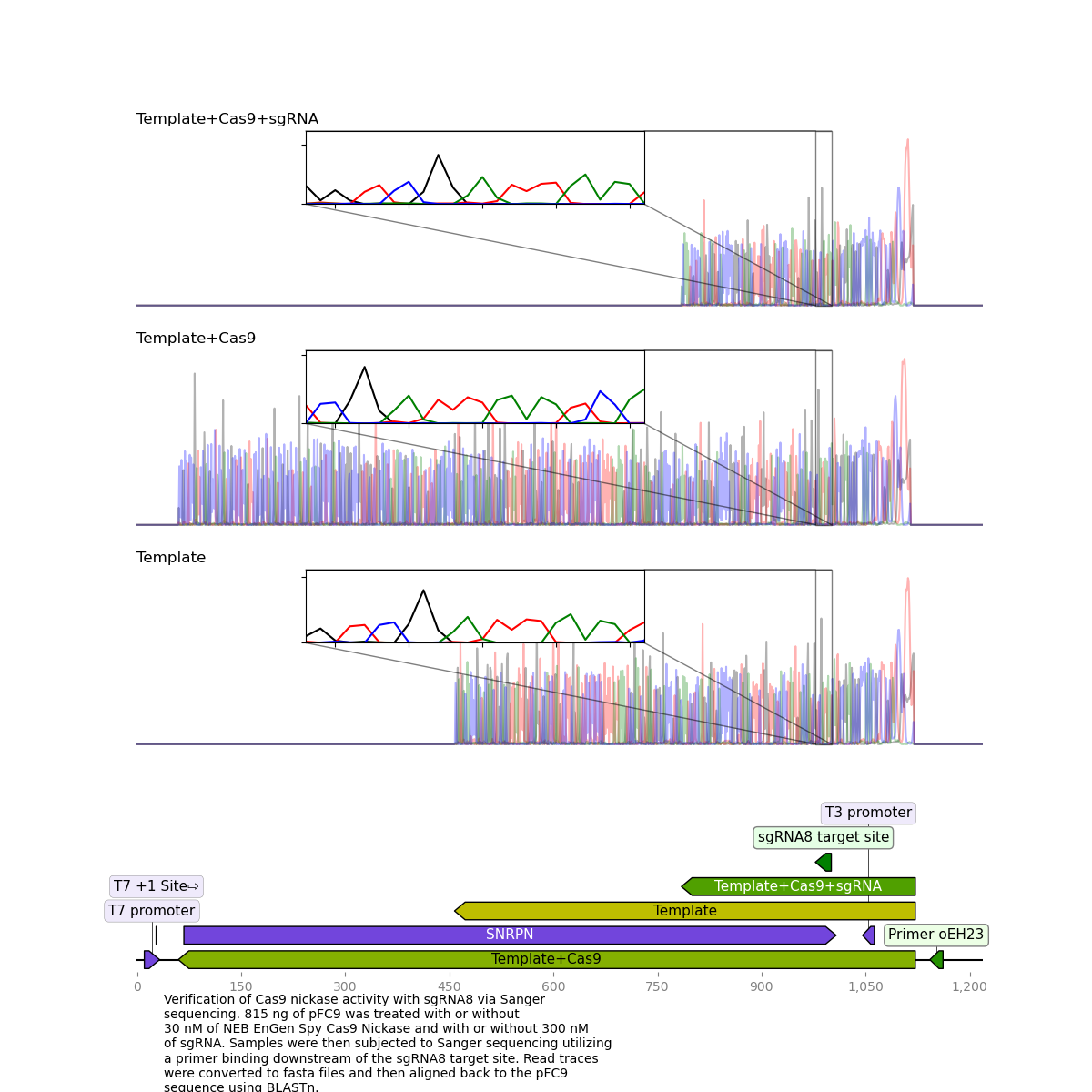
While for sgRNA 8 and all other sgRNAs tested the treated sample reads were much shorter than template + Cas9 or template reads they continue to align well past the sgRNA target site.
I think it will be best to re-run this protocol with just the cas9 treated samples early Monday morning so I can get samples to sequencing that same day. If I see similar nick efficiency on the gel and get similar reads then that will point to an issue at sequencing level.
I talked to Fred about this and showed him the reads. We noticed that there is definitely something going on at target sites. Below are the traces for sgRNA8 reads with and without the guide RNA. SgRNA 8 target site is highlighted in blue in both traces.
Cas9 no sgRNA #
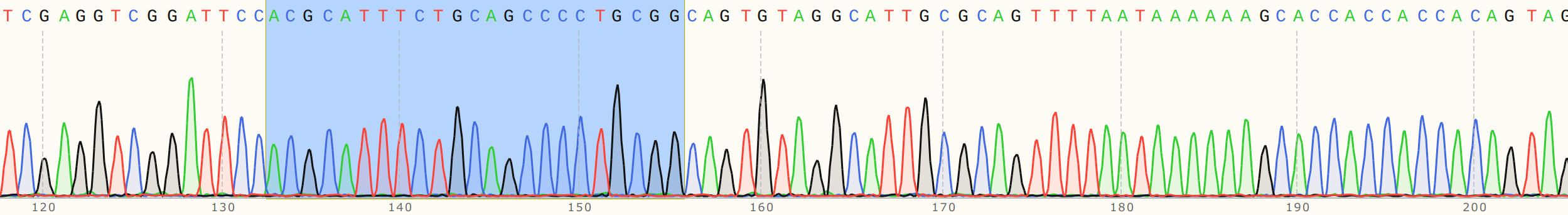
Cas9 + sgRNA #
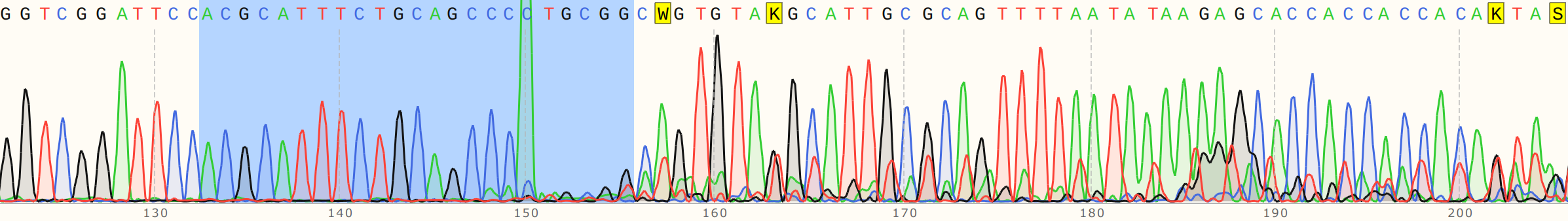
Without sgRNA reads are strong all the way through, but with sgRNA there is a clear drop in signal at the target site, a bit more read that actually aligns and then just noise. Fred suggested that as the polymerase reaches the end of nicked samples if highly palindromic sequences are present the nascent DNA strand can form a secondary structure, binding to itself which produces a 3' that the polymerase can then use as an effective primer. This may explain strong signals after nick sites that do not align back to the template. Contamination with other sgRNAs may also be an issue.
2/13/22 #
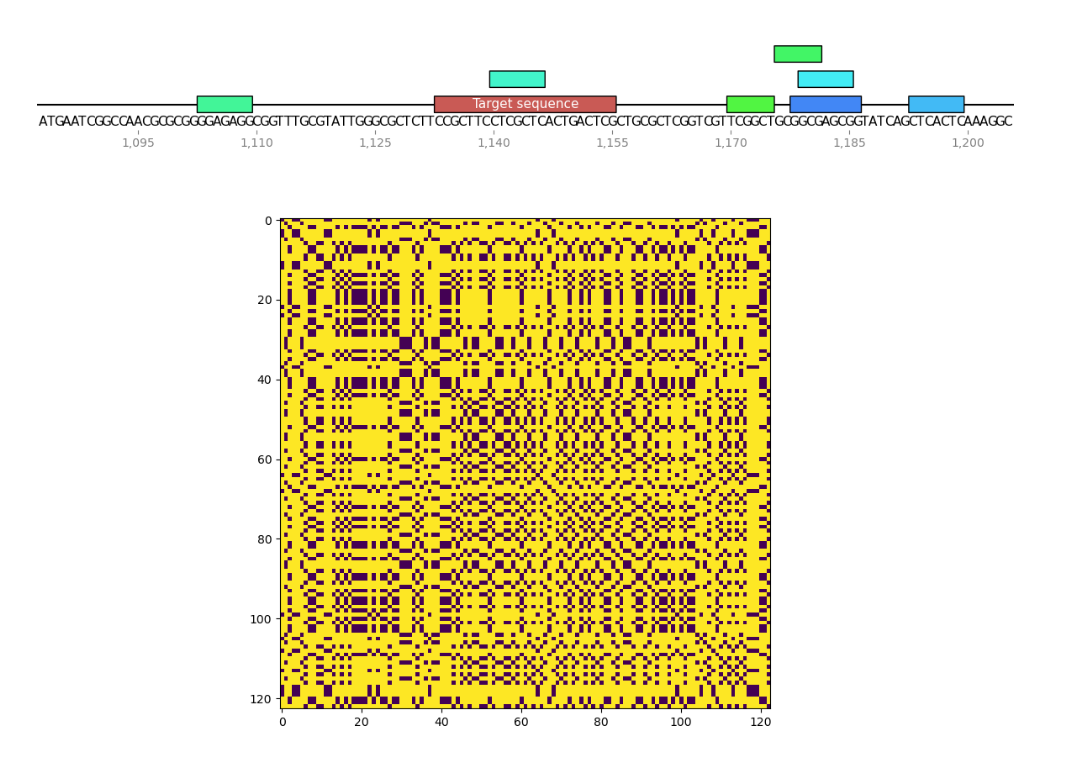
I wrote a small program to find palindromes around Cas9 target sequences and create dotplots of these sequences as well to see if recently tested sgRNAs were significantly more palindromic than sgRNA4. A few of the plots are shown below. Unlabeled features in the sequence map represent palindromes.
Palindrome analysis #
sgRNA 4 #

sgRNA 8 #
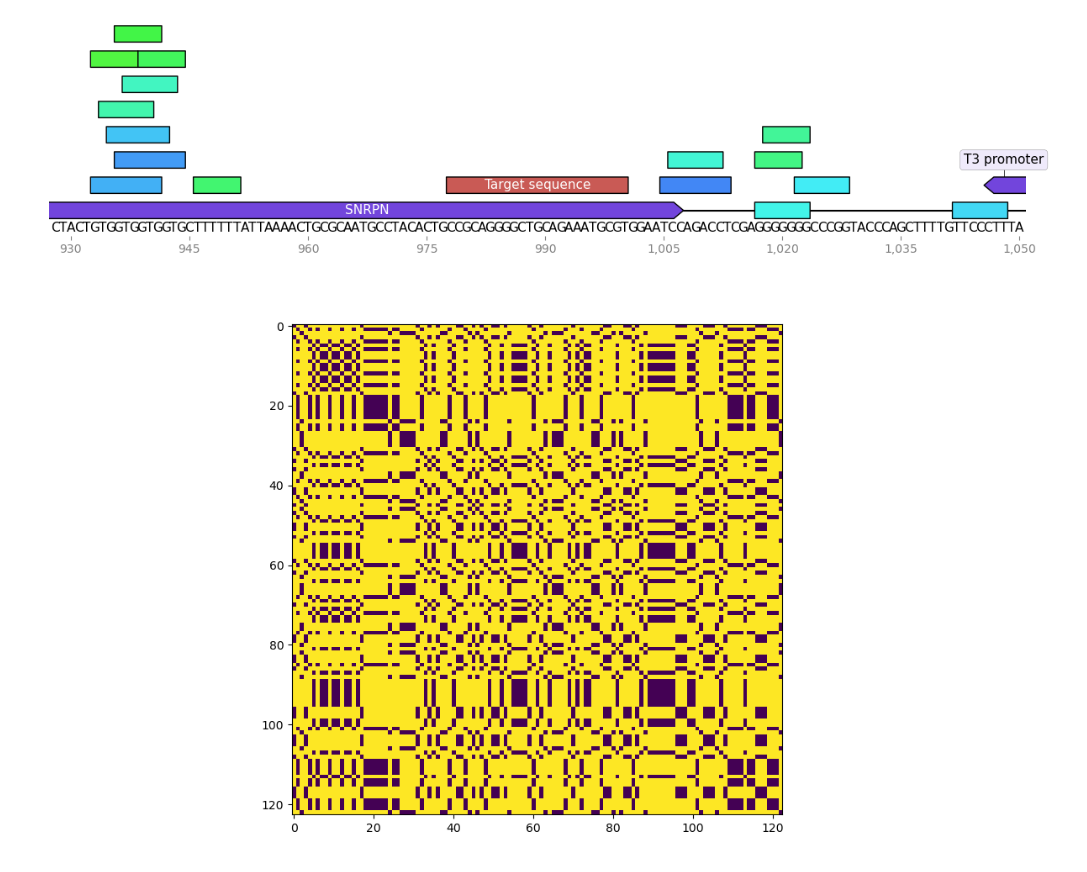
sgRNA 9 #
2/14/22 #
in vitro digestion of pFC9 with sgRNAs 4-9: Follow up to sgRNA 5-9 validation sequencing results #
Digested pFC9 with sgRNAs 4-9 according to NEB EnGen Cas9 nickase in vitro digestion protocols as usual the only modification to the protocol I made was to increase sgRNA concentration to 300 nM. A complete description of the reactions is available at this link.
After completing in vitro digestion protocol sample volumes were increased to 200 ul with npH20. Sample 0 (template only) which had a much larger total volume was split into 200 ul aliquots. I then preformed standard phenol-chloroform EtOH precipitations on all samples using homemade phase lock columns. After completing precipitation I suspended samples in 8 ul 10 mM Tris HCL and combined the previously split sample 0 aliquots. I reserved 2 ul of each sample for analysis on agarose gel and prepared the remainder for sequencing at the DavisSeq center. Sample 0 was split into five samples; one for each sequencing primer used. I then boiled sequencing samples for 5 minutes and then placed immediately onto ice. Samples were then taken directly to the sequencing center.
Analysis by agarose gel #
After dropping off samples I ran the remaining un-boiled 2 ul aliquots of each sample out on an agarose gel which is shown in the image below.
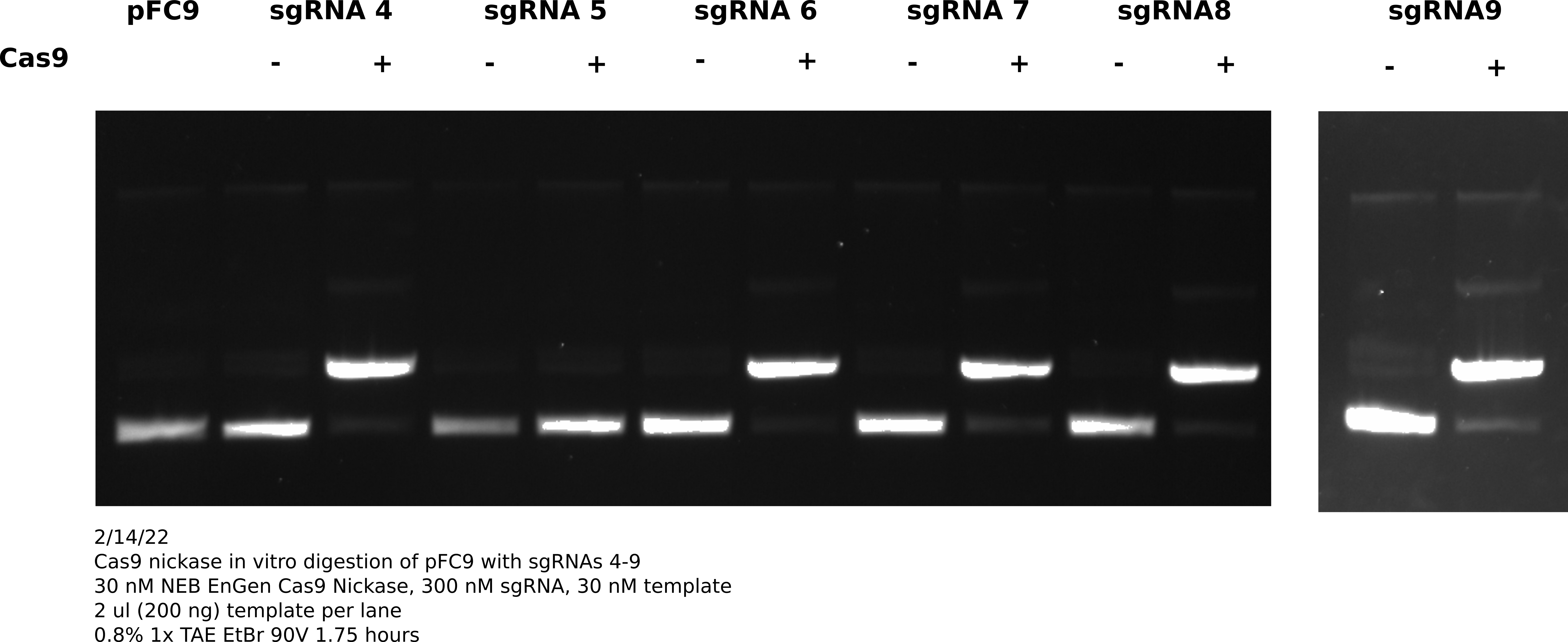
Results are basically as expected. sgRNAs 6-8 show > 90% efficiency while sgRNA 5 shows almost no conversion of supercoiled -> nicked which is reflected results from 2/8/22. Additionally, sgRNA 4 also shows high efficiency (>90%) mirroring results from 1/26/22.
Additionally, this gel shows that no activity is observed with any of the samples when only sgRNA is present.
Literally as I was writing the above sentence I realized what I did wrong. I intended to have a Cas9 with and without sgRNA samples but instead I have sgRNA with and without Cas9 samples. I should still be able to determine if nicking is targeted and access Sanger results but I will likely have to repeat as this is not the gel I would want to show.
Analysis of sequencing results #
Received sequencing results back from Davis center on the 16th. Analyzed them using Casanger workflow; a summary of the results are shown and discussed below. The complete workflow output can be downloaded from this link.
sgRNA 4 #
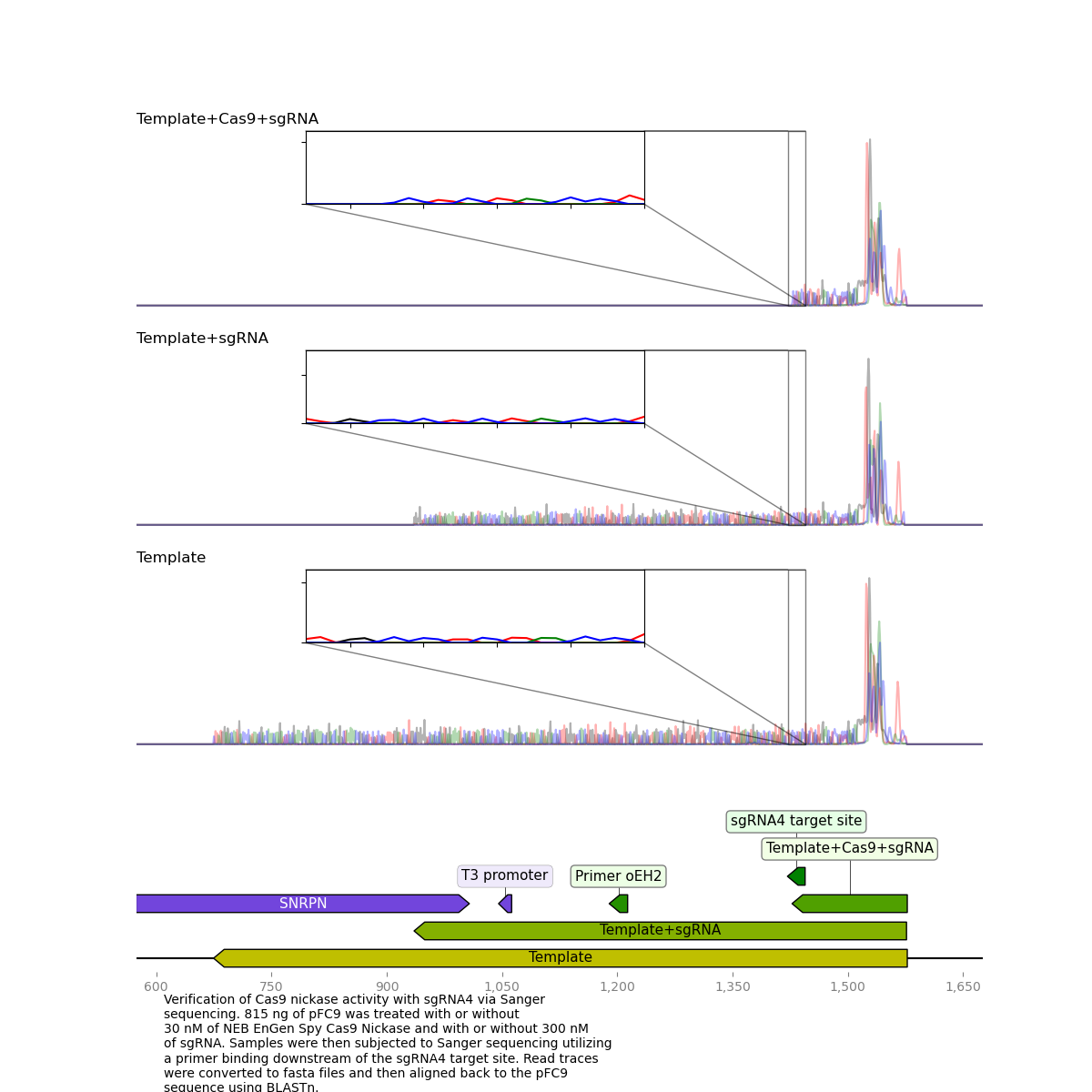
Results for sgRNA 4 look basically as expected, besides from the large spike in signal at the start of the read it is nearly identical to previous results with sequencing dying exactly at the nick target site.
sgRNA 8 #
This sgRNA failed to produce clean killed reads on the last go around but worked very well this time around.
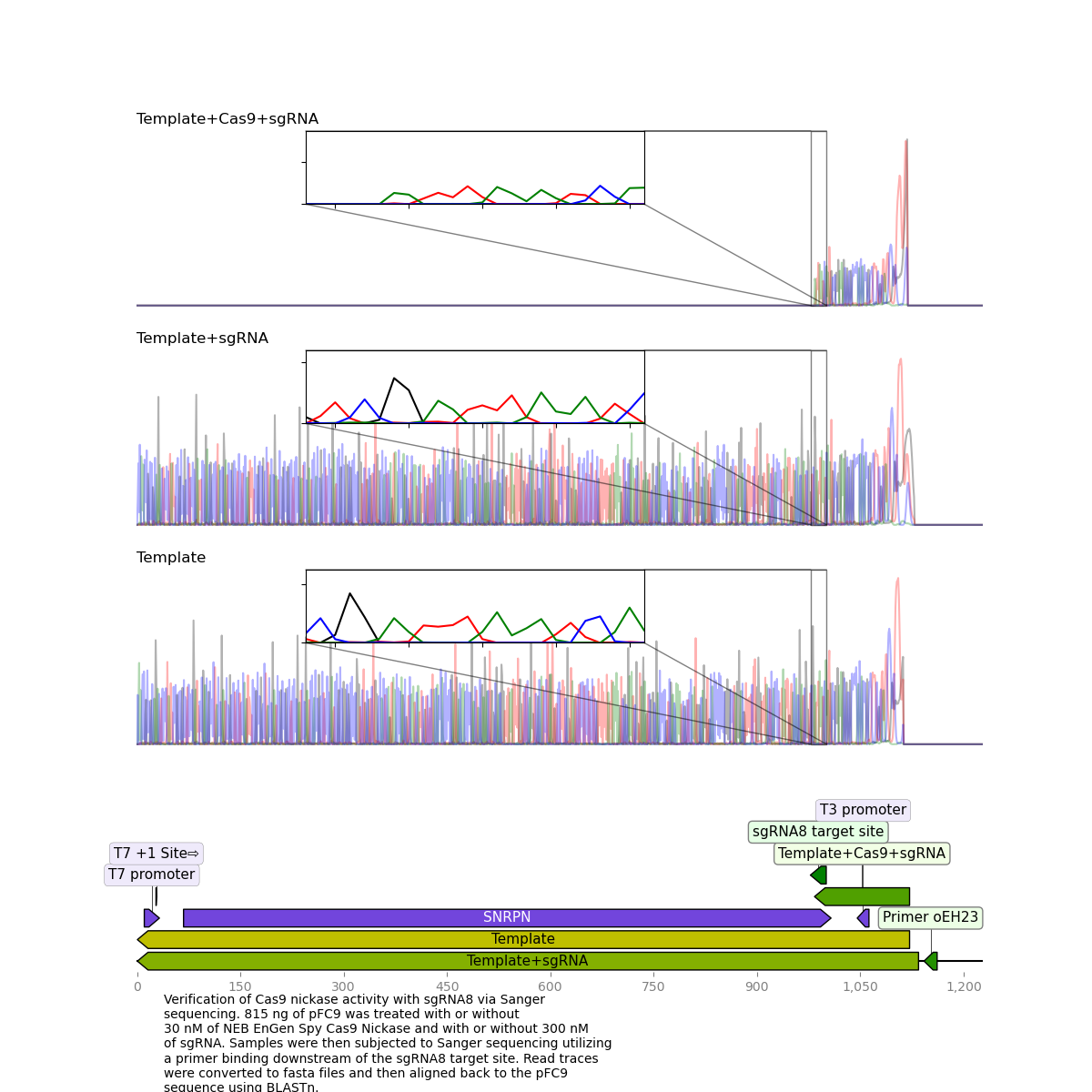
sgRNA 6 #
After looking at sgRNA 4 and 8 you might expect all samples to have worked well, but that was not the case. sgRNA 6 sample read straight through the nick site with the typical drop in signal seen in other nicked reads. This is despite showing nearly identical activity based on the agarose gel.
It is very strange to me that despite similar activity as shown by agarose gel different sgRNAs are producing very different results upon sequencing. To me, this is pointing to some mechanic of sample processing either before or during sequencing causing inconsistencies. After talking to Fred he suggested linearizing templates before boiling to reduce chance of re-annealing. In next test I plan to use HindIII to linearize directly after digestion with Cas9. Then move forwards with ProteaseK digestion and Phenol-Chloroform extraction followed by boiling for 5 mins as usual. It may also be helpful to freeze samples faster by spinning down PCR tubes in cold room and then putting in EtOH dry ice bath.
2/23/22 #
In vitro digestion of pFC9 with Cas9 nickase followed by HindIII digestion #
Prepared a total of 5 samples for Cas9 digestion protocol; all using pFC9 as the template. One sample with untreated pFC9 (no Cas9 no sgRNA), one with sgRNA4 and Cas9 and three replicates of sgRNA6 + Cas9. I am using sgRNA6 as the test condition as it has not shown clean killed reads in any previous assay and sgRNA4 as a positive control as it consistently cleanly kills reads.
Experimental overview #
Only changes I am making to previous Cas9 digestion protocols is following digestion with Cas9 I increase sample volume to 150 ul using npH20, rCutSmart 10X buffer and HindIII-HF. Then allow HindIII to digest samples for 30 minutes. I will remove 50 ng of DNA before and after digesting with HindIII to access Cas9 and HindIII activity on each sample respectively.
Experimental setup #
Tables showing reagent formulations and dilutions are shown in the tables below. The complete spreadsheet with all calculations is available at this link.
After completing Cas9 and HindIII digestions ran all samples on 0.8% 1x TAE agarose gel at 80V for 1 hour. Gel image is shown below.
Agarose gel of samples #
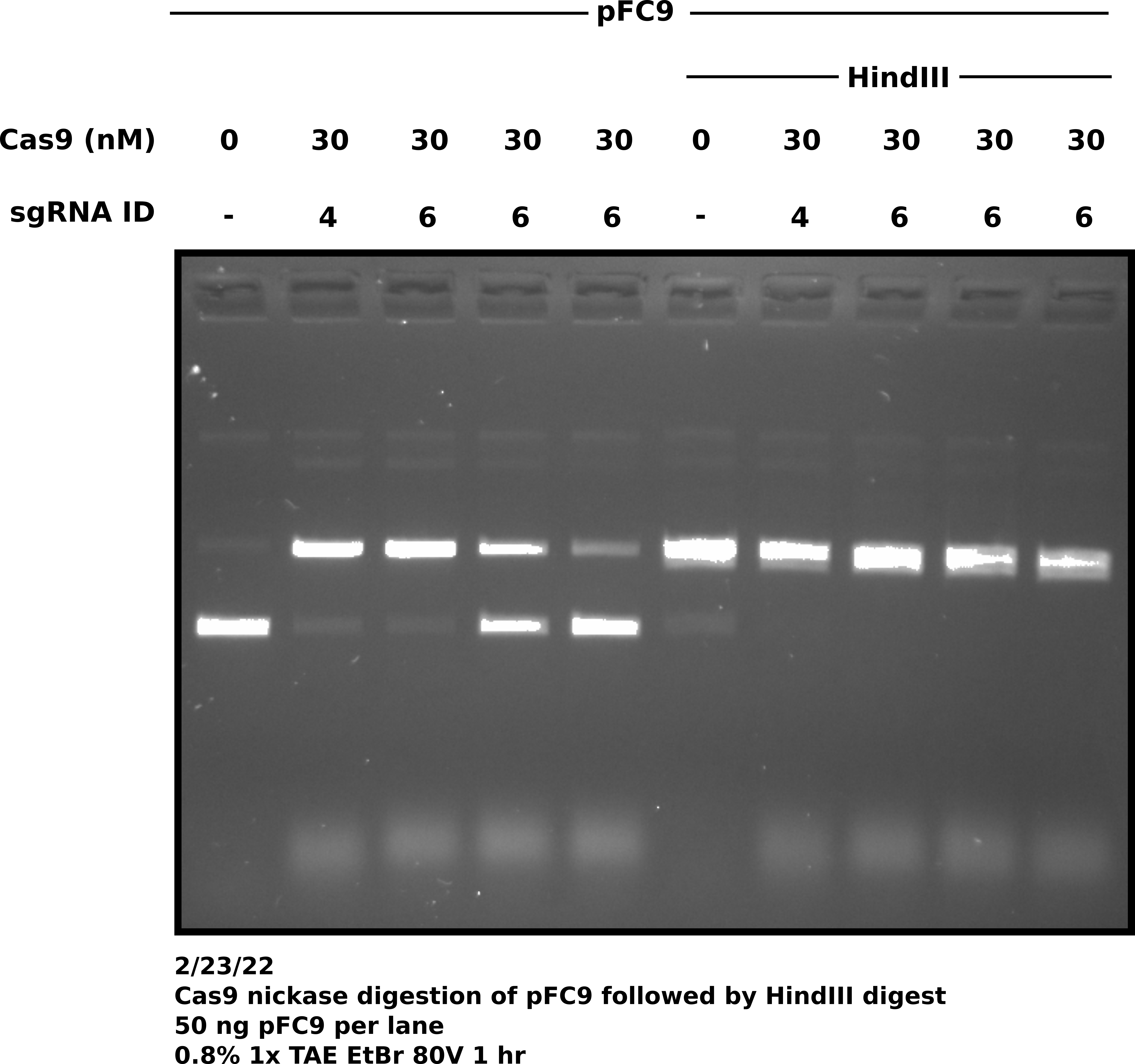
Surprisingly all sgRNA 6 replicates do not show the same degree of digestion. This does not bode great for my consistency but will actually be sort of nice to have for sequencing. I can send 1 replicate that worked well and one that didn’t work so well and compare sequencing results. sgRNA 6 samples were prepared separately (no master mix). I might need to mix dilutions better.
After completing protocol samples were re-suspended in 10mM Tris-HCL and frozen until next sequencing opportunity at Davis center (Friday).
Sanger sequencing submission #
Sample preparation #
On Friday (2/25/22) I prepared 4 samples for sequencing. The untreated pFC9, the sgRNA4 sample and two sgRNA6 samples; replicate 1 and 2. Replicate 1 showed high nicking efficiceny while 2 showed around 50 percent (see agarose gel image lanes 3 and 4 from 2/23/22.)
After thawing samples I boiled in the thermocycler for 5 minutes. After boiling I placed onto ice and quickly (sprinted) to the cold room where I had a PCR tube centrifuge waiting. I spun down samples and then dropped into EtOH dry ice bath to flash-freeze them.
Do not use sharpie to label tubes going into an EtOH bath
I realized my sample labels would be dissolved as soon as the tubes hit the ethanol. Use an EtOH resistant marker or a p1000 pipette tip to float the samples on the surface of the bath and label the top of the tubes. Luckily due to the geometry of the tubes, I was able to tell the order of the samples.
After EtOH bath transfered samples to a cooler with ice pack and dropped off samples at the Davis sequencing center.
Sample submission #
Description of samples sent in the table below. Also can be downloaded at this link.
| Sample Number | Primer Oligo ID | Primer Name | sgRNA | Cas9 | Template | HindIII-HF | Davis Sample Name | Notes |
|---|---|---|---|---|---|---|---|---|
| 1 | oEH2 | pFC8tac_tac_promoter_Primer_2 | NA | FALSE | pFC9 | TRUE | 1 + 2 | pFC9 untreated |
| 2 | oEH24 | oEH19_Cas9_validator | 4 | TRUE | pFC9 | TRUE | 2 + 24 | sgRNA4 sample |
| 3 | oEH2 | pFC8tac_tac_promoter_Primer_2 | 6 | TRUE | pFC9 | TRUE | 3 + 2 | sgRNA6 replicate 1 |
| 4 | oEH2 | pFC8tac_tac_promoter_Primer_2 | 6 | TRUE | pFC9 | TRUE | 4 + 2 | sgRNA6 replicate 2 |
Sanger sequencing data analysis #
Received sequencing results via email. Traces can be downloaded at this link. Analyized results by eye using Snapgene and casanger snakemake workflow.
There are two template reads for plots because a Cas9 + template alone read was not included. Currently the workflow is structured so that it expects three reads per plot. Therefore I just duplicated the template read. It is the exact sample in both thetemplate_1andtemplate_2plots.
Off the bat the template only read looks good with clean signal.
sgRNA6 replicate 1 #
This is the replicate that showed high nicking efficiency on the most recent and previous gels but until now has not had clean killed reads.
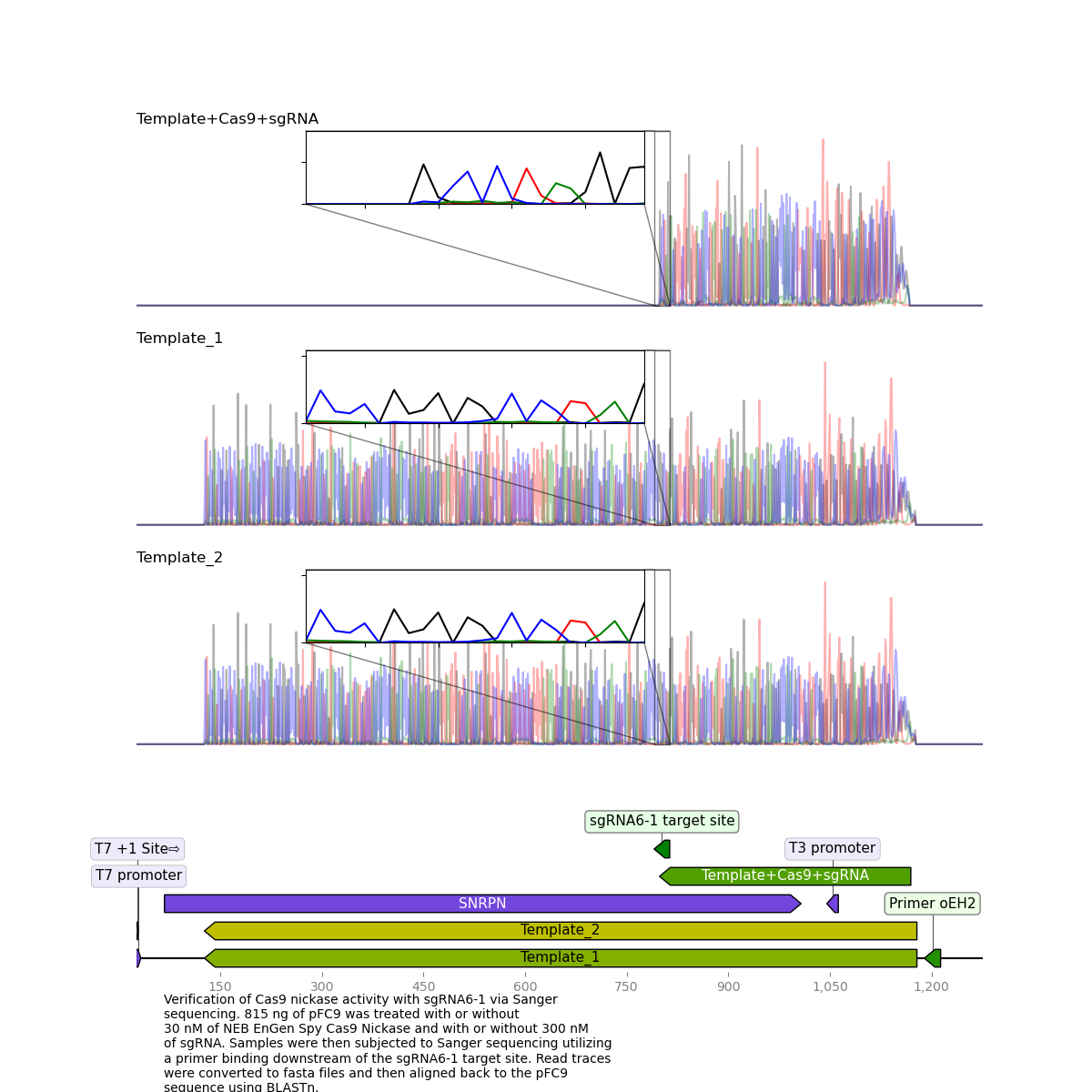
Here, it is clear to see that the read was killed exactly at the target site.
sgRNA6 replicate 2 #
Replicate 2 which did not show high nicking efficiency via agarose gel reflected these results upon sequencing.
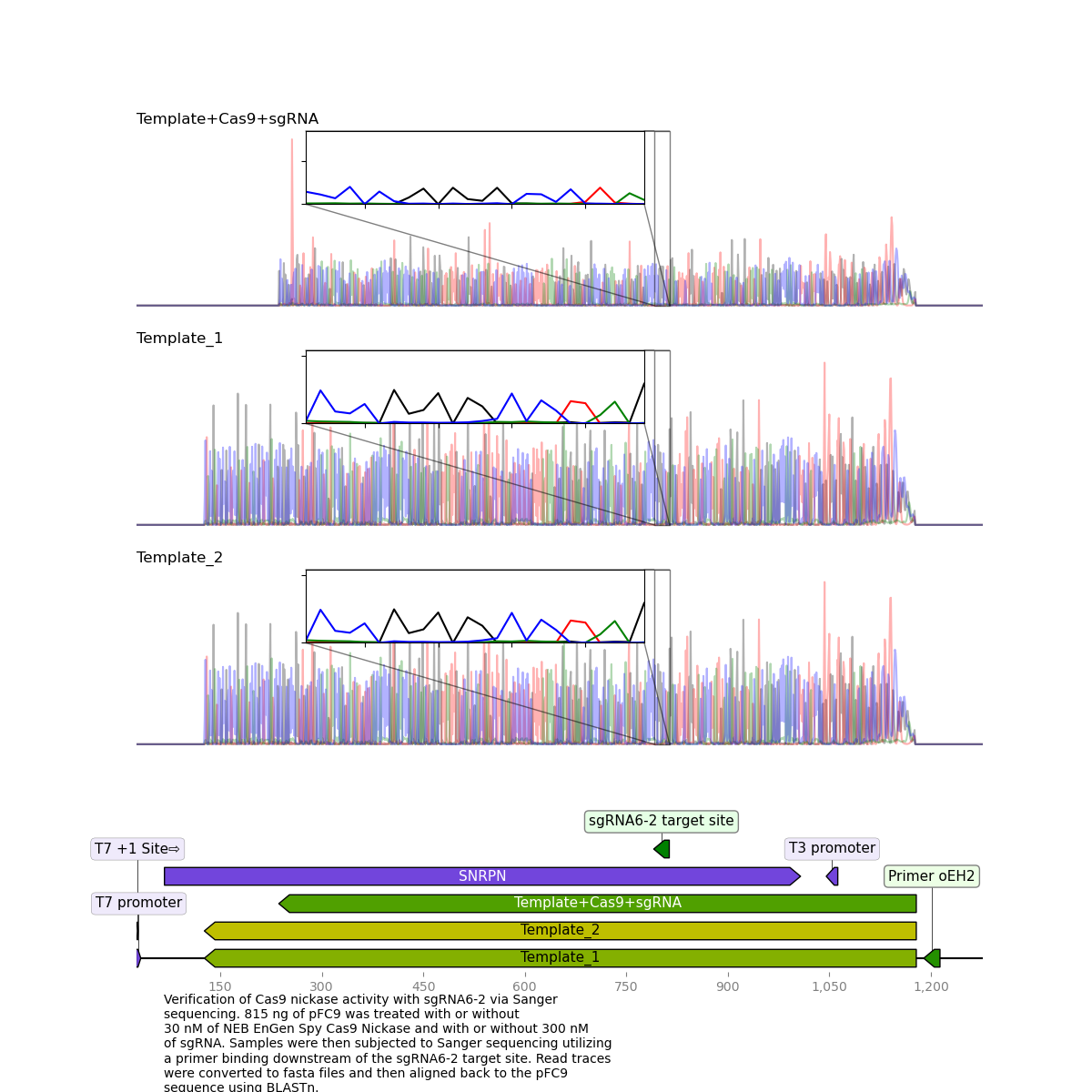
This suggests that a high nicking efficiency is required for proper clean kills of Sanger reads.
I am not including casanger plots of sgRNA4 reads because I used the wrong primer which bound too close to the nick site resulting in a very short read that the pipeline has difficulty aligning via BLASTn.
sgRNA4 #
3/7/22 #
sgRNA 10-12 synthesis #
Using NEB EnGen sgRNA synthesis kit and protocol I synthesized sgRNAs 10-12. sgRNA oligos, IDs and target sites are shown in the table below.
| Name | Oligo ID | Location | Sequence (5' -> 3') | Target Seq (with PAM) | Date created | Date ordered | Date arrived |
|---|---|---|---|---|---|---|---|
| T7Init_10-03-02-2022 | oEH61 | I8 | TTCTAATACGACTCACTATAGCTGCCATAGCCTCCTCGCCTGTTTTAGAGCTAGA | CTGCCATAGCCTCCTCGCCTCGG | 3-2-22 | 3-2-22 | 3-4-22 |
| T7Init_11-03-02-2022 | oEH62 | I9 | TTCTAATACGACTCACTATAGGTCACACCGACTCATCCCCCGTTTTAGAGCTAGA | GTCACACCGACTCATCCCCCTGG | 3-2-22 | 3-2-22 | 3-4-22 |
| T7Init_12-03-02-2022 | oEH63 | I10 | TTCTAATACGACTCACTATAGCCGAGGGAGTGTTTGTTTTAGAGCTAGA | ccgagggagtgtttggg | 3-2-22 | 3-2-22 | 3-4-22 |
Synthesis results #
After completing EngGn kit protocol and phenol chloroform extraction using homemade phase lock columns.
| Name | sgRNA concentration (ng/ul) | Final volume (ul) | Synthesis method | Purification method |
|---|---|---|---|---|
| sgRNA-10 | 1189 | 50 | NEB EnGen sgRNA kit | Phenol chloroform phase lock followed by EtOH precipitation |
| sgRNA-11 | 1342 | 50 | NEB EnGen sgRNA kit | Phenol chloroform phase lock followed by EtOH precipitation |
| sgRNA-12 | 1269 | 60 | NEB EnGen sgRNA kit | Phenol chloroform phase lock followed by EtOH precipitation |
Agarose gel #
After specing samples diluted 1 ul of each sgRNA in 39 ul of TE. Split samples and digested one of each sample with 1 ul RnaseA. Ran whole sample (20 ul) on agarose gel with EtBr to visualize sgRNAs.

Analysis #
sgRNAs are there and are sensitive to RnaseA.
3/8/22 #
Validation of sgRNAs 4, and 6-12 #
Attempting to do a full validation of sgRNAs using updated linearization protcol to potentially use as data for validation compotent of a figure.
Reaction setup #
All samples, concentrations and dilutions are described in this spreadsheet. A summary of the included reactions are shown in the table below. Template used for all samples was pFC9VR18 which was choosen at random from all possible T7Init series plasmids.
| Sample number | sgRNA | Treatment | sgRNA Concentration (nM) | Cas9 Concentration (nM) | Template concentration (nM) |
|---|---|---|---|---|---|
| 4 | sgRNA-4 | Template + Cas9 + sgRNA | 300 | 30 | 3 |
| 6 | sgRNA-6 | Template + Cas9 + sgRNA | 300 | 30 | 3 |
| 7 | sgRNA-7 | Template + Cas9 + sgRNA | 300 | 30 | 3 |
| 8 | sgRNA-8 | Template + Cas9 + sgRNA | 300 | 30 | 3 |
| 9 | sgRNA-9 | Template + Cas9 + sgRNA | 300 | 30 | 3 |
| 10 | sgRNA-10 | Template + Cas9 + sgRNA | 300 | 30 | 3 |
| 11 | sgRNA-11 | Template + Cas9 + sgRNA | 300 | 30 | 3 |
| 12 | sgRNA-12 | Template + Cas9 + sgRNA | 300 | 30 | 3 |
| T+C | NA | Template + Cas9 | 0 | 30 | 3 |
| T | NA | Template | 0 | 0 | 3 |
Reason for BsaI use in digestion post-Cas9 treatment
Previously I have used HindIII and EcoRI. However I am using new sgRNAs (10-12) which cut much closer to the TSS and also happen to be very close to the EcoRI site in T7Init series plasmids. BsaI is a reliable single cutter and its cut site is located a good distance downstream of the furthest sgRNA target site. This ensures that if a read terminates early it is clearly due to Cas9 treatment and not RE digestion.
After completing all reactions samples were phenol-chloroform extracted using homemade phase lock columns and resuspended in 8 uls Tris-HCl and frozen.
Agarose gel of samples #
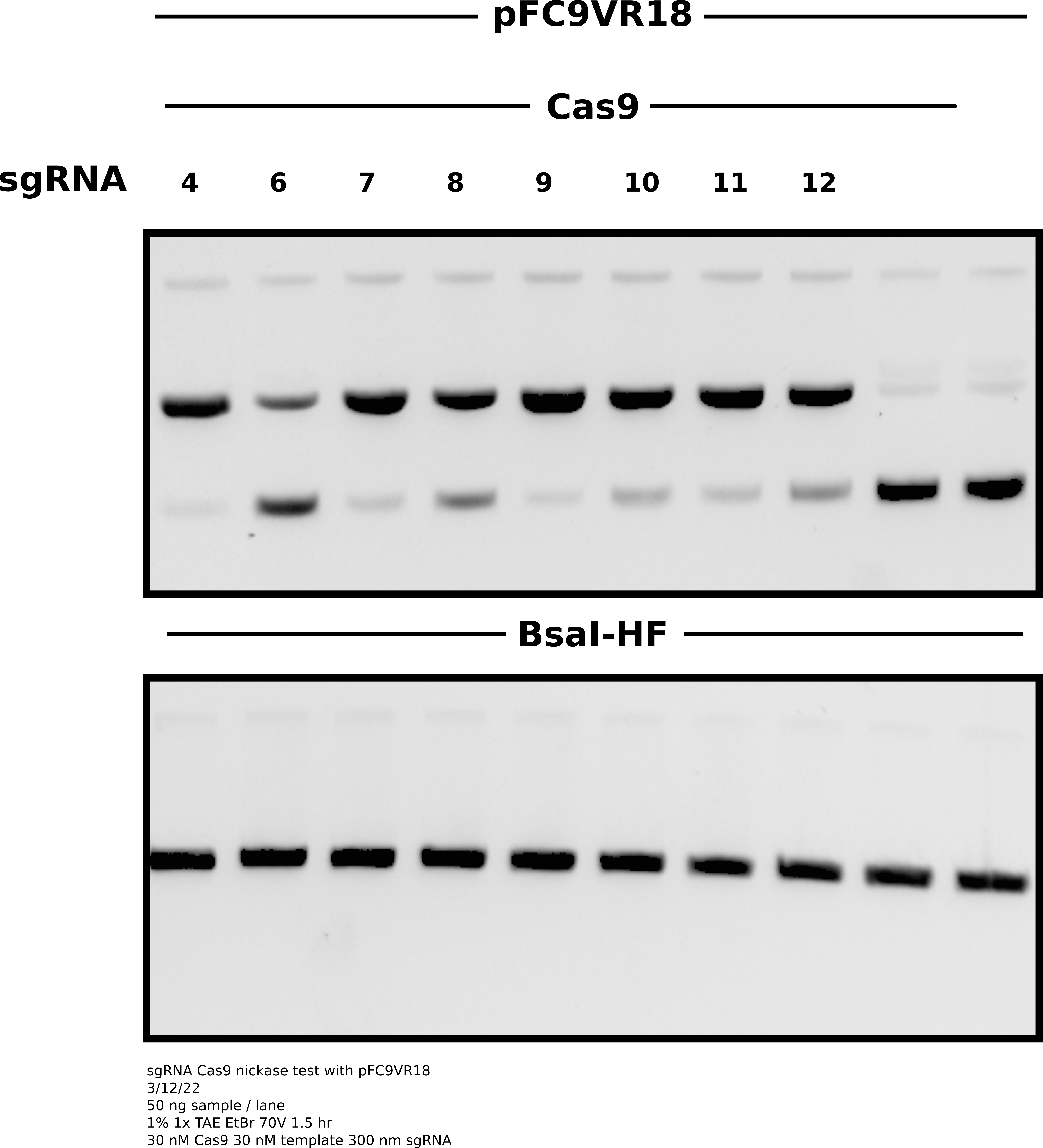
Analysis #
Everything looks really nice except for sgRNA6 which I have previously experinced inconsistencies with. The upside is that all three of the new sgRNAs seem to work which is nice as they target close to the TSS. In the future I will drop sgRNA6 from experiments as it is not consistent enough to include and seems to have a high chance of ruining otherwise nice looking gels.
3/14/22 #
Prepared subset of samples for sequencing at Davis Sanger center according to table below.
| Sample | ID | Primer | Primer ID | Sample prep date | Davis Sample name | Sent for sequencing |
|---|---|---|---|---|---|---|
| pFC9VR18 + sgRNA10 | 1 | oEH21 | p21 | 3/8/22 | 1 + p21 | 3/14/22 |
| pFC9VR18 + sgRNA11 | 2 | oEH21 | p21 | 3/8/22 | 2 + p21 | 3/14/22 |
| pFC9VR18 + sgRNA12 | 3 | oEH64 | p64 | 3/8/22 | 3 + p64 | 3/14/22 |
| pFC9VR18 + Cas9 | 4 | oEH21 | p21 | 3/8/22 | 4 + p21 | 3/14/22 |
| pFC9VR18 | 5 | oEH21 | p21 | 3/8/22 | 5 + p21 | 3/14/22 |
Before sending samples boiled for 5 mins, spun down in cold room and froze in EtOH dry ice bath. After flash freeze placed samples in -80C before walking over to sequencing center with samples on ice at 11 AM.
Sanger sequencing results and analysis #
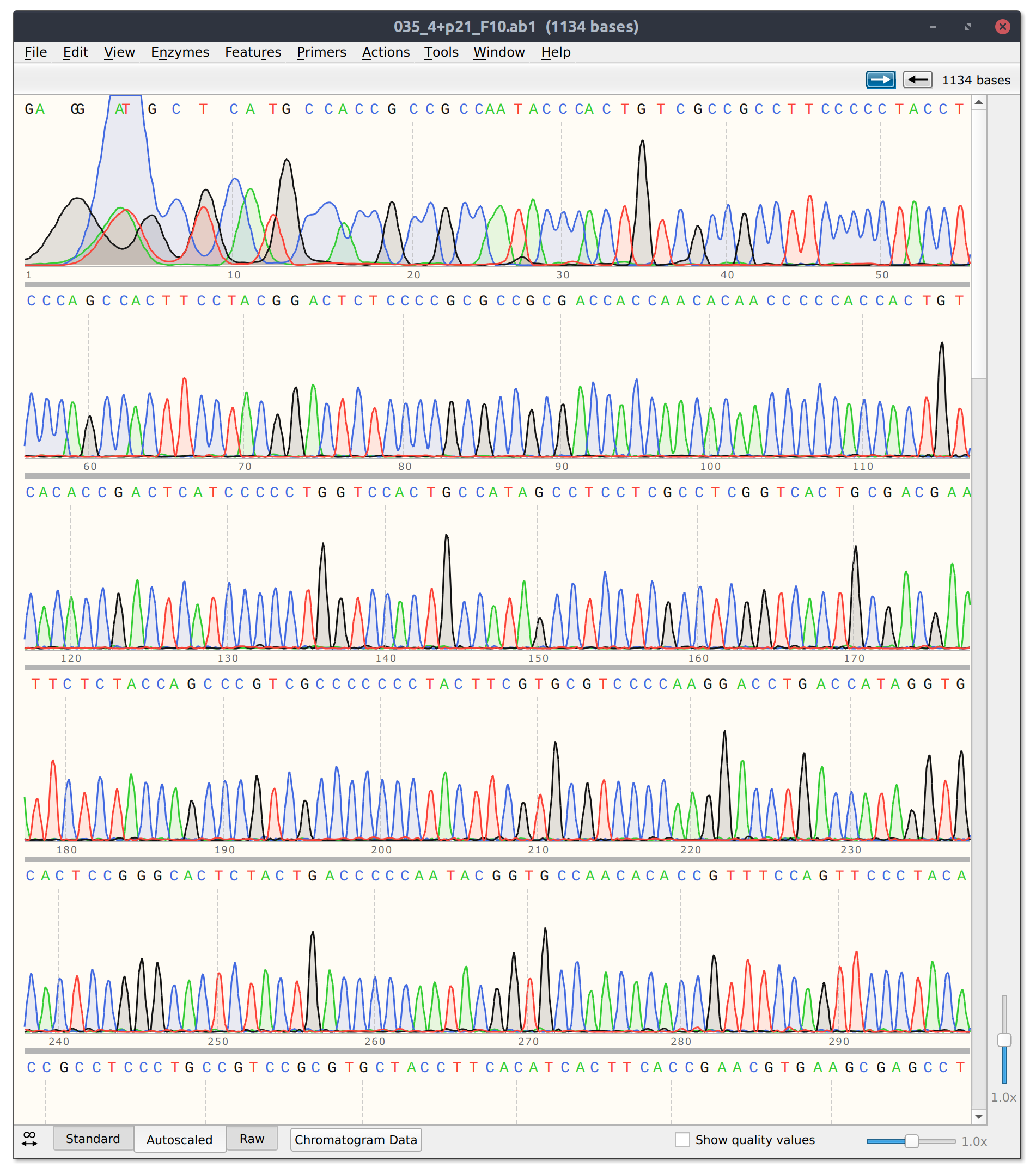
Sanger sequencing results were returned Wednesday 3/16 and can be downloaded at this link.
The template only and Cas9 + template control reads look good. Trace from template + Cas9 shown in the image below.
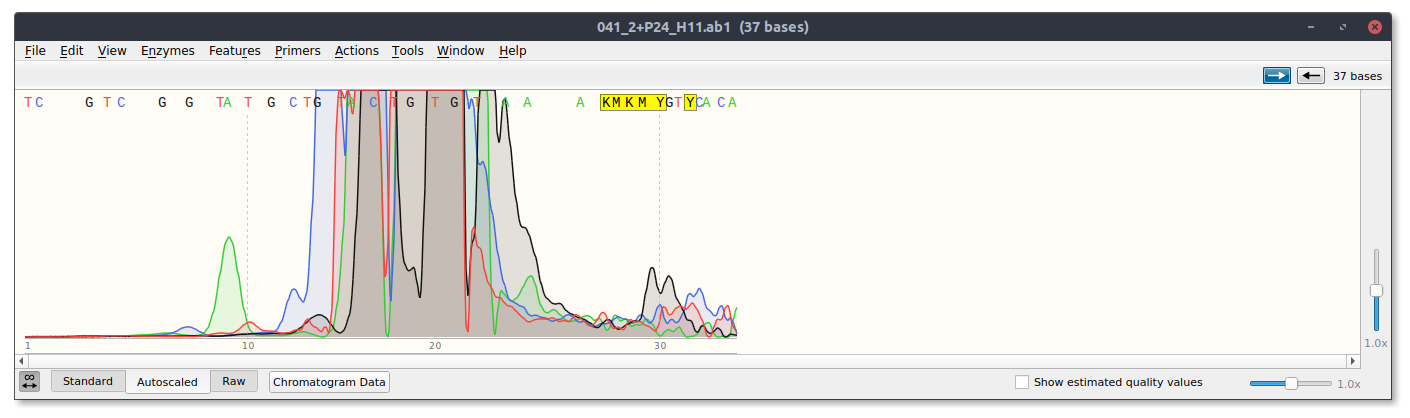
However, for sgRNA reads there is good and bad news. The bad is that sgRNA12 (closest to TSS) and sgRNA 10 did not seem to kill their reads. Traces shown below with target sites highlighted.
sgRNA 12 trace
sgRNA10 trace
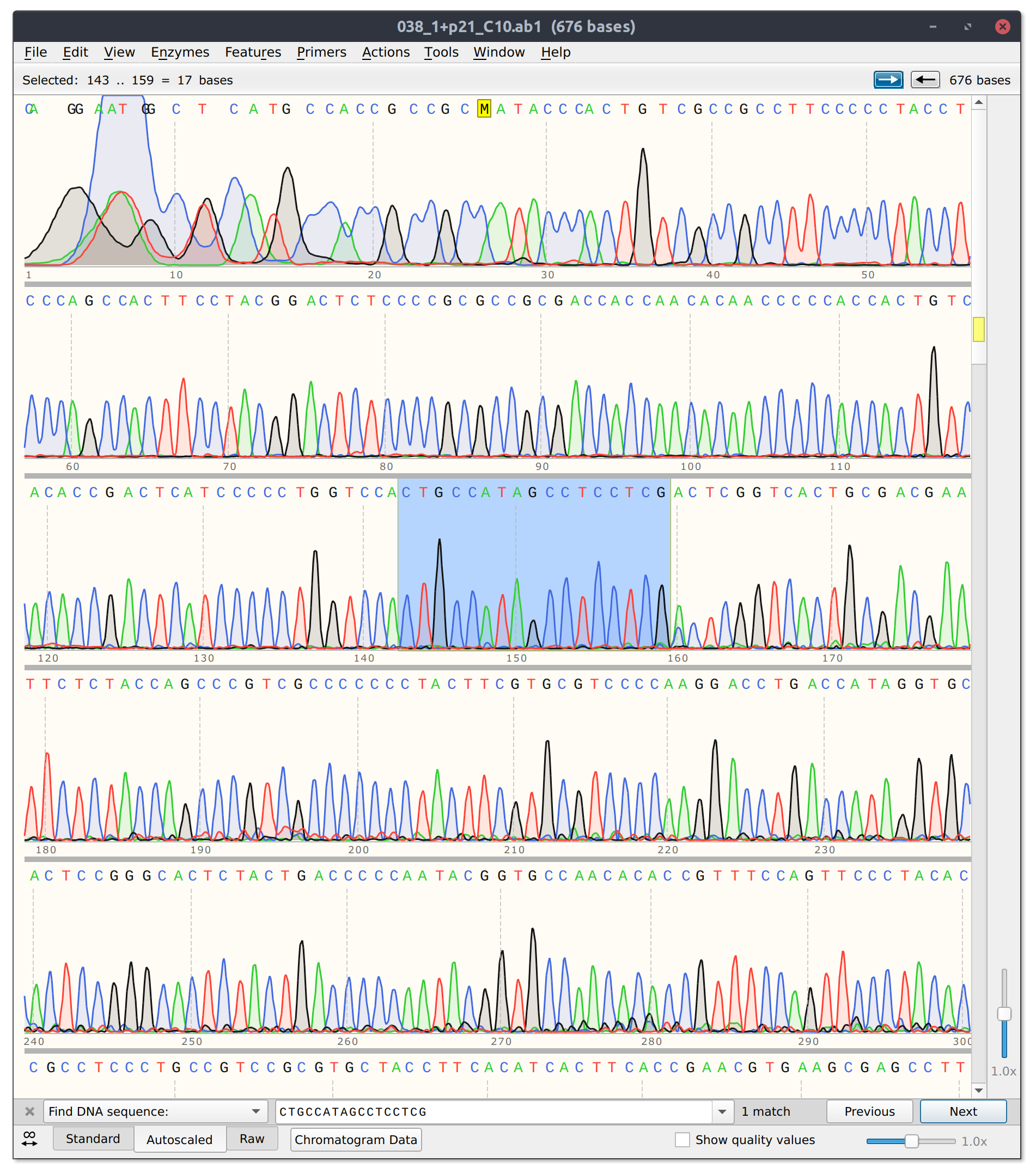
This is frustratingly again despite very clean results on the agarose gel.
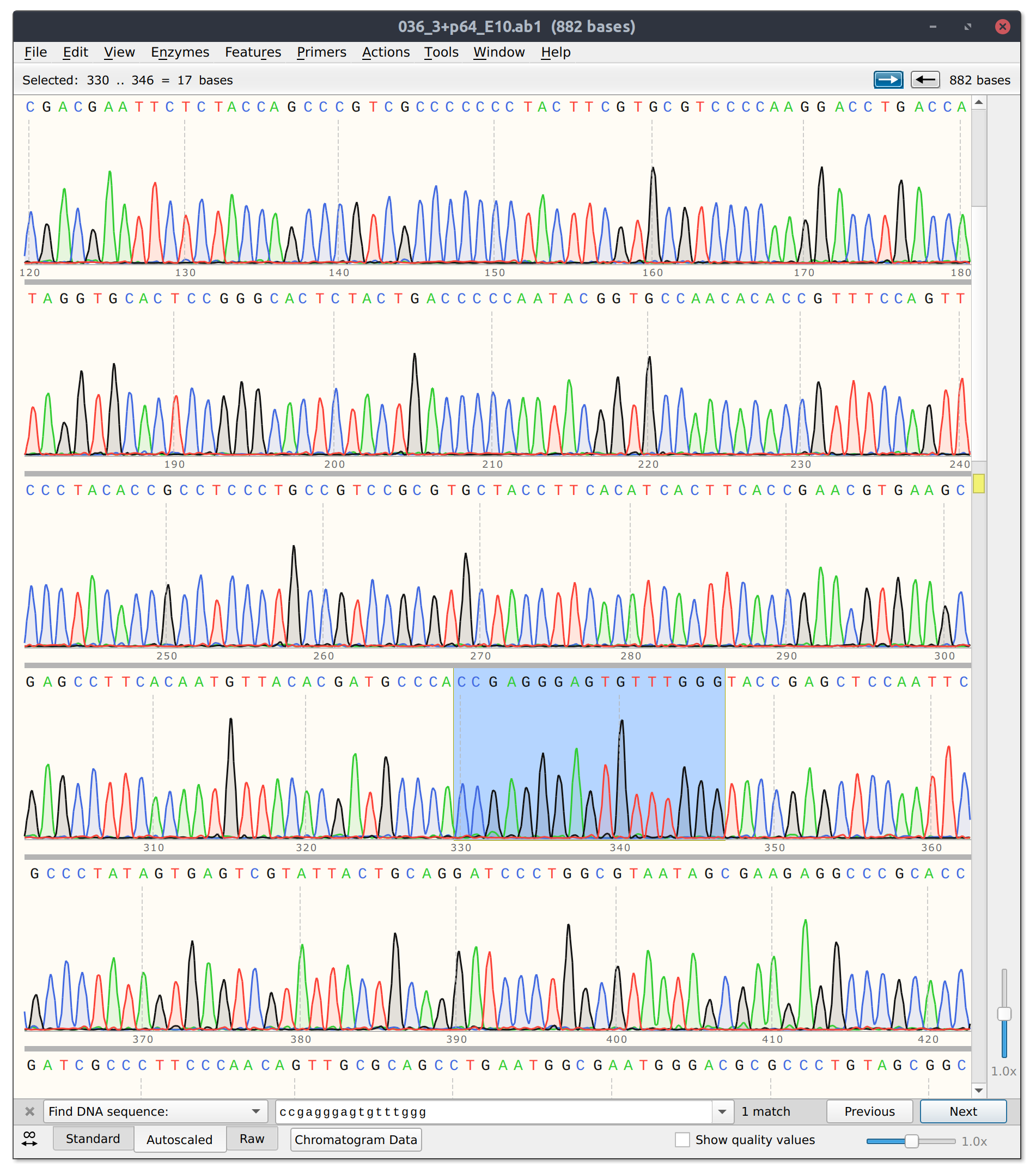
The good news is that sgRNA11 which is basically co-located with sgRNA10 did seem to effectively kill its read. Trace with as much of target site present highlighted below.
sgRNA 11 trace
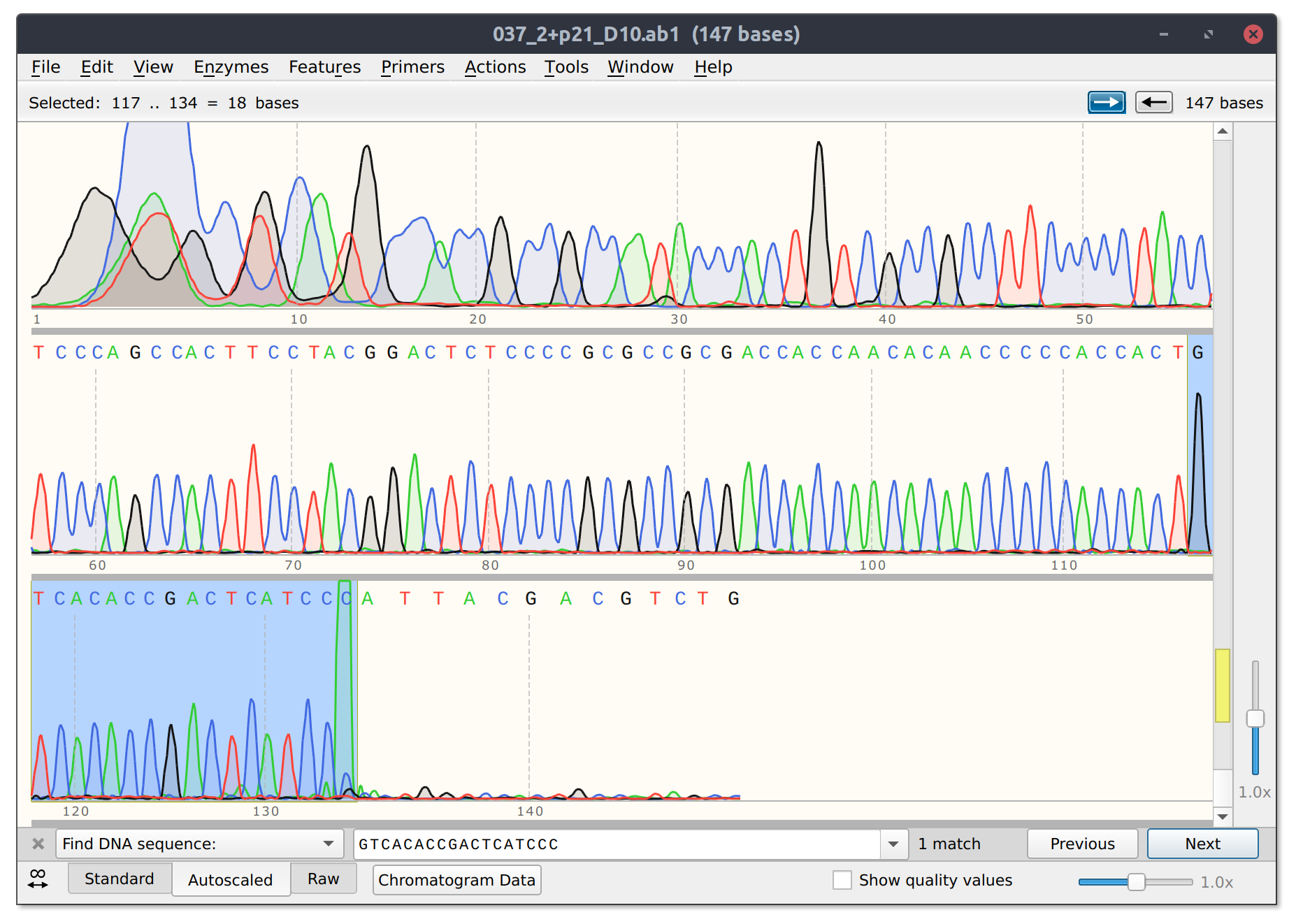
Analysis #
There are two ways to look at these results.
The methodology is not robust enough to return consistent results.
Inconsistencies between gel and sequencing results can be explained by the fact that Sanger sequencing is a poor way to resolve nicks. This is supported by inconsistent results with some sgRNAs, namely sgRNA 6 and the fact that very well nicked substrates often do not produce cleanly killed reads upon sequencing.
The methodology is robust, inconsistencies between gels and sequencing results indicate off target effects.
sgRNAs that produce a high fraction of nicked template but do not produce consistent clean sequencing results are because of off-target effects of the sgRNA. Off target nicks would still shift molecules to the same relaxed height but when sequenced reads would not terminate at the target site. This is supported by the fact that some sgRNAs produce very consistent results both on the gel and when iterogated by sequencing; namely sgRNA4.
This is also supported by the fact that nicking endonucleases (BspQI) which presumably would have little to no off target nicking produce cleanly terminating reads.
I think the best way to test this is to do many replicates of a BspQI treatment and send for sequencing. If everything comes back terminated then it will be safe to assume the method is robust. In the future should probably do this kind of more rigourous testing before exititly jumping into a new method.
The experiment described above was carried out on 3/17/22 - 3/18/22 and is documented in Nick Validation: BspQI sanity check.