Another day, another contaminated PCR sample #
OD9/23 PCR grid reactions #
See reaction setup table for mapping samples to reagents.
| Sample | ng/ul |
|---|---|
| MM | 833.9 |
| H20 | -24.7 |
| 1 | 1009 |
| 2 | 372.9 |
| 3 | 1034 |
| 4 | 1000 |
| 5 | 241 |
| 6 | 254 |
| 7 | 25.7 |
| 8 | 1346 |
| 9 | 1346 |
| 10 | 291 |
| 11 | 1191 |
| 12 | 133.7 |
| 13 | 312 |
| 14 | 1787 |
| 15 | 1205 |
| 16 | 1239 |
| 17 | 1059 |
| 18 | -31 |
| 19 | -32 |
| 20 | -23 |
| 21 | 73.3 |
| 22 | -30 |
| 23 | 1676 |
| 24 | 230.7 |
| 25 | 118.4 |
Contamination seems to be spreading to other reagents beyond just the dNTPs as I first thought was the issue. Wiped down all pipettes with DnaseAway which should destroy Dnases and DNA molecules that might be present on the pipettes. Then set up the PCR reactions described below.
Pre and post pipette clean PCR #
Testing if cleaning pipettes removed contaimination issues by using new reagents and setting up reaction before and after cleaning with DnaseAway. Since amplifications occured when only single reagents were used in the previous PCR test I am testing single reagent reactions and using reactions with only H20 as the control.
- H20 is np Dnase / Rnase free aliquots
- “Fresh” refers to opened for first time for this reaction
| Sample | H20 | Pipette | Reagent | Pipette Cleaning |
|---|---|---|---|---|
| 1 | Fresh npH20 | p20 | Deepvent | Pre-DnaseAway |
| 2 | Fresh npH20 | p20 | red dNTPs | Pre-DnaseAway |
| 3 | Fresh npH20 | p20 | Daisy dNTPs | Pre-DnaseAway |
| 4 | Fresh npH20 | p20 | Forward primer | Pre-DnaseAway |
| 5 | Fresh npH20 | p20 | Rev primer | Pre-DnaseAway |
| 6 | Fresh npH20 | p20 | Lab Taq | Pre-DnaseAway |
| 7 | Fresh npH20 | p20 | H20 | Pre-DnaseAway |
| 8 | Fresh npH20 | p20 | H20 | Pre-DnaseAway |
| 9 | Fresh npH20 | p20 | Deepvent | Post-DnaseAway |
| 10 | Fresh npH20 | p20 | red dNTPs | Post-DnaseAway |
| 11 | Fresh npH20 | p20 | Daisy dNTPs | Post-DnaseAway |
| 12 | Fresh npH20 | p20 | Lab Taq | Post-DnaseAway |
| 13 | Fresh npH20 | p20 | Forward primer | Post-DnaseAway |
| 14 | Fresh npH20 | p20 | Rev primer | Post-DnaseAway |
| 15 | Fresh npH20 | p20 | H20 | Post-DnaseAway |
| 16 | Fresh npH20 | p20 | H20 | Post-DnaseAway |
9/23 PCR grid reactions #
Ran some of products from 9/23 PCR tests out on a 0.8& agarose gel in 1x TAE buffer with 1ul / ml EtBr for 45 mins. Lane layout is listed in the table below. Refer to 9/23 PCR grid test sample table for mapping sample numbers to reagent treatments.
Lane layout #
| Sample | Lane |
|---|---|
| MM | 1 |
| 2 | 2 |
| 4 | 3 |
| 7 | 4 |
| 9 | 5 |
| 11 | 6 |
| 12 | 7 |
| 17 | 8 |
| 21 | 9 |
| 23 | 10 |
Gel image #
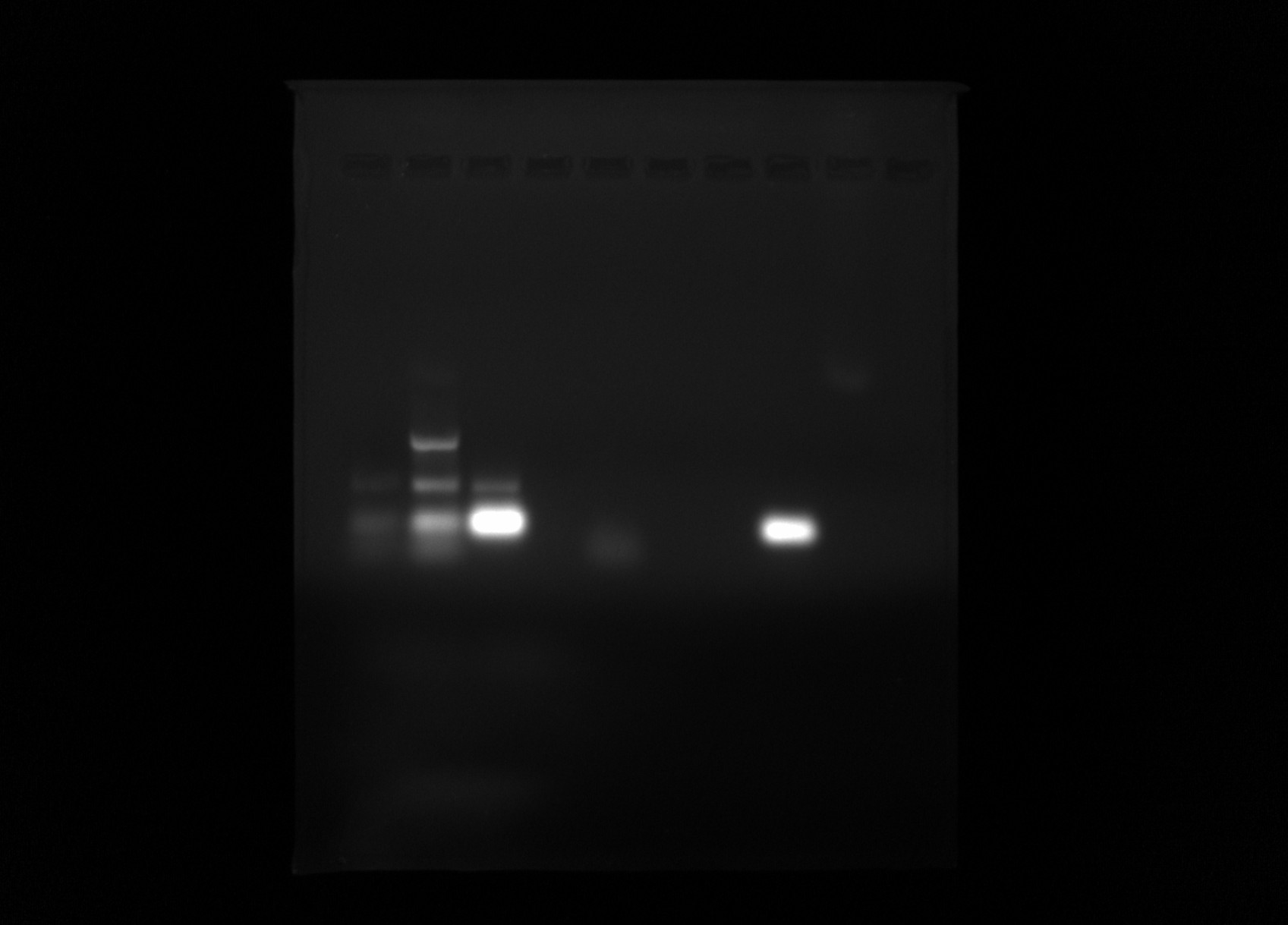
Lane 6 looks like no contaimination which is sample 11 (new buffer, Daisy dNTP, deepvent taq but with no primers) so should not be amplification anyway. Lane 8 (sample 17: original Go, Lab taq, Daisy dNTP and both primers) shows a very strong band. This may be primer dimer though.
IVT with Gyrase #
Given the somewhat surprising results of 9/22/21 IVT that included Gyrase samples I wanted to do more replicates with more inserts. So in order to condense the number of reactions I would need to do I did not include gyrase RnaseH controls.
Here I followed standard IVT lab protocol but added 6ul of 5x Gyrase buffer to each sample (substituting for 6ul H20) and then pretreated one fraction of plasmid with DNA gyrase for 30 mins at 37C. After Gyrase treatment samples were transcribed with T7.

Overall Gyrase treatment did not really seem to make a difference for any of the inserts. Granted I did not heat kill Gyrase after treatment so really testing R-loop formation in the presence of DNA gyrase instead of just on a hyper supercoiled template.
Testing DNA Gyrase activity #
Given that some VR insert plasmids do not seem to change R-loop formation patterns when pre-treated with DNA gyrase I wanted to test for activity of the enzyme. So I came up with the basic protocol shown below.

If Topo is working this should cause relaxation which Gyrase should then restore back to the supercoiled state.
Reaction setup #
- 600 ng pFC9 (2 ul)
- 3 ul 10x rCutsmart (Topo buffer)
- 6 ul 5x Gyrase buffer
- 19 ul npH20
Take 20 ul from initial mix and treat with 1ul Topo for 30 mins at 37C. Then take 10ul of the topo treated fraction and treat with 1 ul DNA Gyrase under those same conditions.
Gel image #
After completing both reactions ran all three samples out on 0.8% TAE agarose gel in 1x TAE buffer for 45 mins at 120V.
| Lane | Sample |
|---|---|
| 1 | Control |
| 2 | Topo treated |
| 3 | Gyrase + Topo treated |
Gel was loaded with 6x purple loading dye.

If anything this is almost the opposite of what I was expecting to occur. Gyrase should not be shifting the band upward. Although Fred brought up the fact that I did not heatkill enzymes after each treatment (which I should have thought to do) and so there could have been competition between them. This is not really a valid assay. Despite this I think that something might be up with these enzymes.