Re-purification of Gibson fragments #
Since pFC9 fragment purification on Friday yielded no DNA, I am guessing this was a significant problem in the last assembly attempt. So today I am starting from scratch and attempting to make high quality pFC9 large fragment fractions to use in the next Gibson assembly attempt.
Check VR fragments #
Before next assembly to be safe I am running the VR fragments out on a gel to make sure they are also there as well.

Ran gel with 100bp ladder 45 mins TAE at 120 V shows no evidence of DNA in any appreciable amounts for these inserts. Need to probably just remake PCR / BglII digestion products before next assembly.
OD of PCR products after adding dNTPs (oops) #
| Insert | ng/ul |
|---|---|
| 6 | 114.6 |
| 7 | 123.6 |
| 8 | 129 |
| 9 | 87 |
| 12 | 123 |
| 15 | 109 |
| 16 | 151 |
| 21 | 66 |
| 23 | 342 |
| 26 | 340 |
| 27 | 303 |
| 28 | 220.9 |
Yields are as expected, much greater. Should not that last couple samples with very high yields looked like some of the reaction had evaporated reducing the volume.
Test transformation effic.eny of found chemically competent cells #
Found some additional what looks like Invitrogen chemically competent E. coli cells in the -80C freezer. There are quite a lot of samples but are unlabeled and I am guessing are very old. To test the transformation efficiency I followed NEB protocol since I do not have remaining Invitrogen recovery broth but protocols are basically the same and with 30 pg of pUC positive control plasmid DNA provided with the NEB kit. I then plated 50ul of the transformed cells and left to incubate overnight in the 37C hotroom.
Fragment PCR and BglII digestion #
Given results of the last gel I used a pre-prepared PCR grid with primers for VR inserts to amplify inserts 6, 7, 8, 12, 15, 16, 17, 18, 19, 21, 22, 23, 24, 25, 26, 27, 28, 30 and 31. After amplification I digested fragments originally cloned as vectors with BglII; Inserts 8, 12, 15, 16, 17, 21, 23, 26, 27, 28, 30 and 31. The reaction components are shown in the table below.
| Reagent | Volume (ul) |
|---|---|
| PCR product | 25 |
| BglII | 0.5 |
| 10x 3.1 buffer | 5 |
| H20 | 19.5 |
After 1hr at 37C in the hot room I aliquots 4ul digested samples and 2ul undigested samples for agarose gel. Also took 3ul of the digested samples and repeated the VR insert PCR protocol without adding additional reagents.

Ran 45 min 0.8 argarose gel at 120V with 100 bp ladder. Many fragments did not show PCR products. Maybe an issue with the PCR grid lacking one or more reagents? Going to repeat the PCR with a fresh made master mix and see if that solves the problem. Based on these results I am redoing samples 6, 7, 8, 9, 12, 15, 16, 21, 23, 26, 27 and 28 using the master mix recipe below.
| Reagent | Volume (ul) |
|---|---|
| Lab Taq | 3 ul |
| OneTaq Buffer | 75 |
| Fwd primer | 7.5 |
| Rev primer | 7.5 |
| H20 | 247.5 |
I then amplified these fragments following standard PCR protocol with total volume of 25 ul per reaction and ~10 ng of DNA and then measured the OD of each sample after amplification.
OD of PCR with fresh master mix #
| Insert | ng/ul |
|---|---|
| 6 | 56.8 |
| 7 | 68.2 |
| 8 | 73.8 |
| 9 | 67.6 |
| 12 | 76.9 |
| 15 | 57.2 |
| 16 | 59.3 |
| 21 | 53.3 |
| 23 | 111.1 |
| 26 | 111.4 |
| 27 | 111.1 |
| 28 | 99.1 |
At this point I realized that I forgot to add dNTPs to the PCR master mix. So I added 0.5 ul dNTP to all samples are re-ran the PCR again without adding any additional reagents. It was approaching 5:30 at this point and so I set up the reaction and let it sit overnight.
Analysis of9/10/31 Sanger results #
First I aligned all sequences to the reference sequences for all inserts and plotted the results as a heatmap shown below.
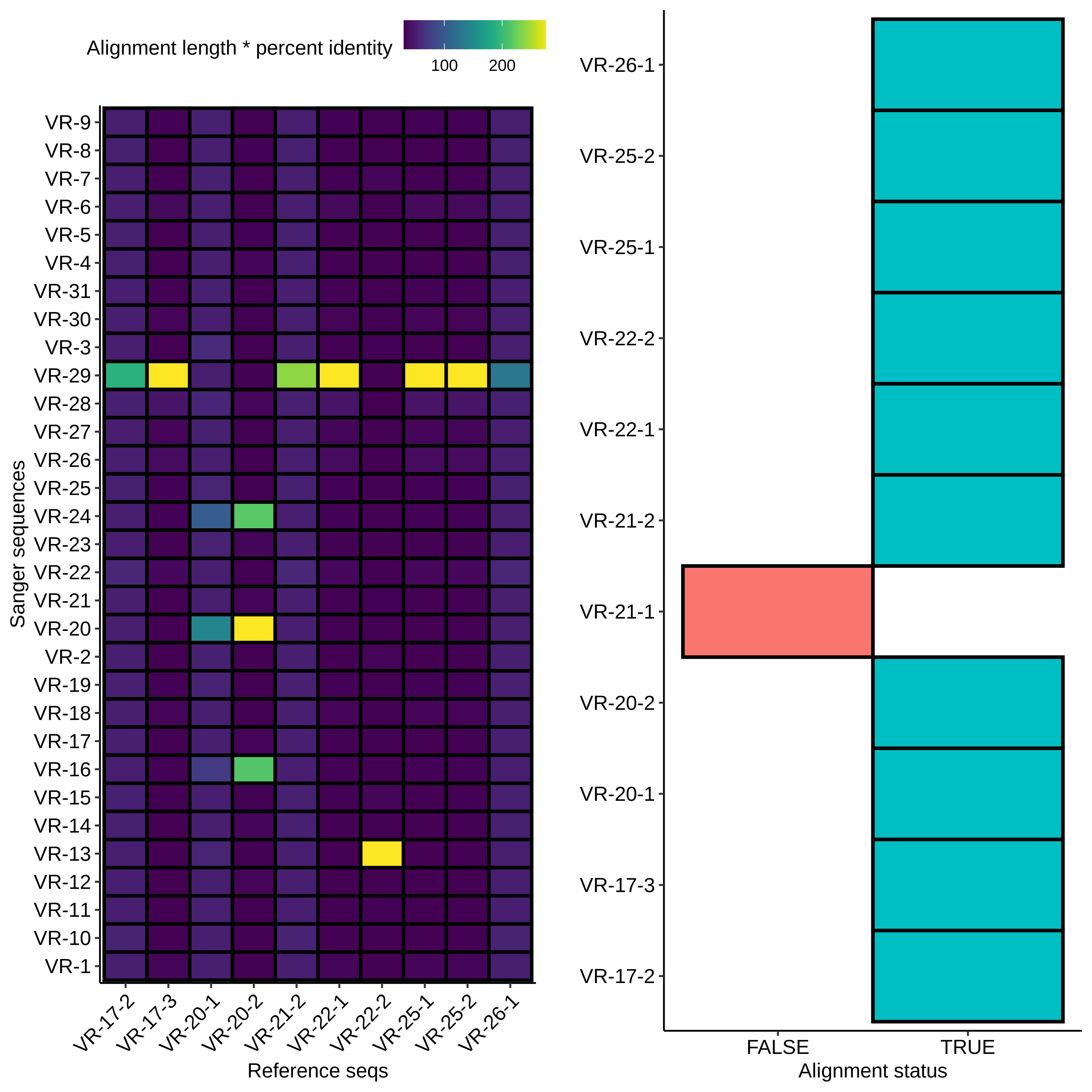
Right away it looks like there was contamination as most of the
samples are mapping to VR-29 (which wasn’t actually part of this
round of assemblies?) but there also appears to be some
additional salvageable material. Midi VR-22-2 clearly maps to
VR-13 and midi VR-20-2 looks like it maps to VR-20. If we look at
alignments with VR-20-2 and alignments between all insert
reference sequences we can see that the hits on VR-24 and VR-16
are due to preexisting homology and not due to contamination.

Additionally the VR-20-21 looks clean and does not indicate
contamination.